来源:BioArt
程序性细胞死亡(programmed cell death, PCD)可以去除无用的细胞以及感染或具有潜在肿瘤性的细胞,在体内平衡,宿主防御病原体,癌症和一系列其他病理学中具有重要作用。目前已经发现的PCD包括细胞凋亡,细胞坏死和细胞焦亡;不同的PCD通过不同的分子机制引导细胞走向不同的命运。
近期的一系列研究表明,这些PCD途径非常灵活,其分子调控具有相当程度的可塑性;例如,尽管炎症性Caspase在诱导细胞焦亡中起主要作用,但炎症性Caspase可以诱导细胞凋亡,相反,细胞凋亡刺激也可引发细胞焦亡。有趣的是,这种灵活性在细胞对感染的反应中最为明显,而细胞凋亡是生物体阻止癌症发展的主要细胞死亡途径。
在这篇综述中,我们总结了不同类型PCD的分子机制,以及这些途径参与生理和病理过程的内在关联性,重点是感染和癌症。尽管不同的PCD途径既可以看作是单一的细胞死亡过程,但在特定的生理条件下,这些PCD可以协调发挥作用,彼此之间存在代偿性的调节。
引言
多细胞生物体的发育和稳态不仅取决于细胞受控增殖,还取决于如何清除异常的细胞,避免对生物体构成潜在的危险。这包括去除有致瘤风险的受损细胞或被微生物劫持用于病原体复制的细胞。程序性细胞死亡(Programmed cell death, PCD)是生物体消除这些异常细胞的主要手段[1-3]。它可以通过机体发育和应激等信号诱导激活,刺激膜结合蛋白和胞质蛋白,通过复杂的转录级联响应和蛋白翻译后修饰触发细胞死亡。
过去三十年的研究中发现了几种不同类型的PCD:细胞凋亡是相对“温和”的细胞死亡方式,一般不会引起免疫或者炎症反应,而细胞焦亡和necroptosis是指相对“剧烈”的细胞死亡,其特征在于细胞膜的破裂和由此产生的炎症诱导因子的释放,包括病原体及其不同的裂解成分,如抗原(图1)。
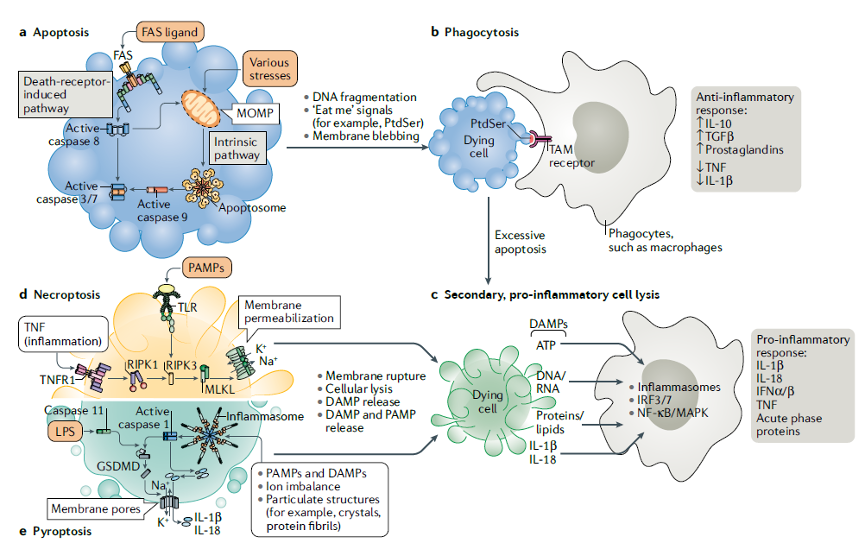
图一:细胞内存在多种方式的程序性细胞死亡(PCD)
不同PCD具有不同的分子机制和死亡方式。细胞凋亡可以由外在(死亡受体诱导)或内在(线粒体或BCL-2调节)途径触发,注:死亡受体也可以参与细胞凋亡的内在途径。死亡受体途径依赖定位于细胞膜上死亡受体与其相应配体的结合(例如FAS受体和其配体),导致Caspase-8的活化。内在途径可以由大量不同的应激刺激诱导,包括DNA损伤或生长因子阻断,以及机体发育过程中的信号刺激。内源性细胞凋亡与线粒体外膜通透有关,线粒体膜通透后导致细胞色素c的释放引起凋亡小体形成和Caspase-9的活化。
两种凋亡途径激活的Caspase(8/9)水解并活化下游效应Caspase-3/7,后者切割数百种细胞蛋白,从而导致核小体间的裸露DNA片段化和细胞膜内侧的磷脂酰丝氨酸暴露于垂死细胞的细胞膜外测等过程。此外,还会引起肌动蛋白重组,导致膜泡化形成凋亡小体。
总的来说,这些caspase诱导的过程加速了细胞的分解。b |表面暴露的磷酯酰丝氨酸被相邻巨噬细胞上的TAM受体识别,进而在任何细胞内容物暴露于细胞外环境之前清除凋亡细胞。PtdSer的识别还可触发吞噬细胞的抗炎反应,介导IL-10、TGF TGF(转化生长因子- TGF)和前列腺素的分泌,以及抑制促炎细胞因子(包括肿瘤坏死因子(TNF)和IL-1杀伤因子)的产生。c |在短时间内大量细胞凋亡,可能会超过吞噬细胞的清除能力,导致所谓的次生坏死,即细胞溶解死亡,细胞内物质从死亡细胞暴露到吞噬细胞,从而加剧炎症。d、坏死性凋亡和细胞焦亡是PCD的主要促炎裂解形式。它们与细胞裂解有关,在感染病原体的细胞的情况下,细胞裂解导致损伤相关分子模式(DAMP)以及病原体相关分子模式(PAMP)的释放。DAMP和PAMP被邻近的吞噬细胞识别,导致促炎细胞因子的产生。通过TNF受体1(TNFR1)或Toll样受体(TLR)刺激(和某些其他信号)诱导坏死性凋亡,引起受体相互作用的丝氨酸-苏氨酸激酶RIPK1和RIPK3的激活。这导致MLKL的构象变化和活化,然后MLKL易位至细胞膜并在细胞膜上打孔诱导膜破裂(d部分)。
细胞焦亡是响应感染而触发的关键PCD途径,其中病原体衍生的产物促进炎性小体的形成,进而激活Caspase-1。此外,据报道脂多糖(LPS)直接刺激Caspase-11(人类中的Caspase-4和Caspase-5)的活化。Caspase-1和Caspase-11都能切割gasdermin D(GSDMD),释放其N-末端片段组装成细胞膜中的孔,导致膜完整性丧失和细胞裂解。Caspase-1还将促炎细胞因子IL-1β和IL-18的前体形式蛋白水解加工成它们的生物活性形式,其通过GSDMD诱导的膜孔释放(部分e)。IFN,干扰素;IRF,干扰素调节转录因子;MAPK,丝裂原活化蛋白激酶。
不同的PCD形式协调应对不同的环境变化,细胞凋亡保证成熟生物体的正常发育和组织稳态,而焦亡和坏死则保护宿主免受病原体和其他外部威胁[4]。对不同PCD途径的遗传敲除小鼠的分析也支持这一观点。在凋亡缺陷的小鼠,比如缺乏抗凋亡因子 bcl 家庭成员BAK,BAX和BOX的小鼠,通常在胚胎发育阶段或者出生后不久死亡, 少数存活到成年的个体表现出明显的淋巴样细胞累积和某些上皮组织未被清除而引起的病理特征(例如爪蹼和阴道闭眼)[5, 6]。
相反,缺乏焦亡或坏死的小鼠出生时健康,但对某些病原体或其他外部损伤的反应有缺陷[7-9]。与这些发现相反,现在有越来越多的证据表明,不同的PCD通路在多个层面上是相互联系的,或许应该考虑在不那么离散的条件下。与这些发现相反,现在有越来越多的证据表明,不同的PCD通路在多个层面上是相互联系。在本文中,我们详细讨论关于不同类型的PCD在健康和疾病中的角色的变化。
PCD途径可大致分为导致细胞裂解和非裂解形式的细胞死亡。裂解类型的细胞死亡,如焦亡和坏死,是由质膜中形成孔洞或其他破口引起的[4]。在裂解型细胞死亡过程中,细胞完整性的破坏是由水的涌入、膜电位的丧失和细胞肿胀引起的,最终导致细胞破裂。
因此,细胞内内容物和(如果存在的话)细胞内病原体的物质——包括RNA、DNA、蛋白质和脂质——都被释放到细胞外空间,作为损伤相关分子模式(DAMPs)和病原体相关分子模式(PAMPs)发挥作用。这些危险信号刺激邻近巨噬细胞和其他临近细胞表达的模式识别受体和其他受体系统,触发促炎细胞因子的释放[10](图1)。非裂解形式的PCD,如细胞凋亡,其特征是将死亡的细胞协调分解成更小的片段,即所谓的凋亡小体,确保细胞内内容物的隔离,包括阻尼(和PAMPs,如果存在)。巨噬细胞主要通过TAM受体识别凋亡细胞,因此将这些死亡细胞清除被认为可以防止炎症(图1)。
细胞死亡方式
细胞凋亡
细胞凋亡过程是由caspases介导的。正常情况下这些天冬氨酸特异性半胱氨酰蛋白酶作为无活性酶原形式存在,在整个进化过程中高度保守[21]。各种上游细胞死亡信号[22]导致Caspases介导的级联活化,引起细胞的形态学和生化变化, 例如:DNA酶介导[23]的DNA片段化,染色质浓缩和膜起泡,凋亡小体等[24]。
生长因子或营养物质阻断,与组织重构或细胞粘附丧失相关的发育信号,类固醇激素或用不同细胞毒类药物刺激等均可触发内源性细胞凋亡(图2a)。内源性细胞凋亡途径涉及BH3促凋亡家族蛋白的表达和活化[25, 26],结合并抑制抗凋亡家族蛋白导致其失活当细胞内所有促存活的BCL-2蛋白在功能上被BH3-only蛋白中和时,释放BAK和BAX以寡聚化导致线粒体外膜破裂,从而诱导线粒体外膜透化(MOMP)[27, 28]。MOMP引起几种线粒体蛋白的释放,其中一些因子能够驱动细胞凋亡过程中下游关键信号传导。
例如,细胞色素c在易位到胞质中时与凋亡肽酶活化因子1(APAF-1)结合,促进凋亡小体的形成。启动子Caspase-9单体,无酶活性的前体形式被募集到凋亡小体中,导致其切割和活化,从而促进效应Caspase-3和7的下游蛋白水解活化[29]。XIAP(凋亡的X连锁抑制剂)是凋亡蛋白(IAP)的抑制剂之一,可以减弱这种caspase级联反应的激活。XIAP可以引发某些Caspase-进行蛋白酶体降解[30]。MOMP还导致SMAC(Caspase-的第二线粒体激活剂,也称为DIABLO)和HTR2(HTRA丝氨酸肽酶2)的释放,它们都可以阻断XIAP从而阻止其抑制Caspase[31]。
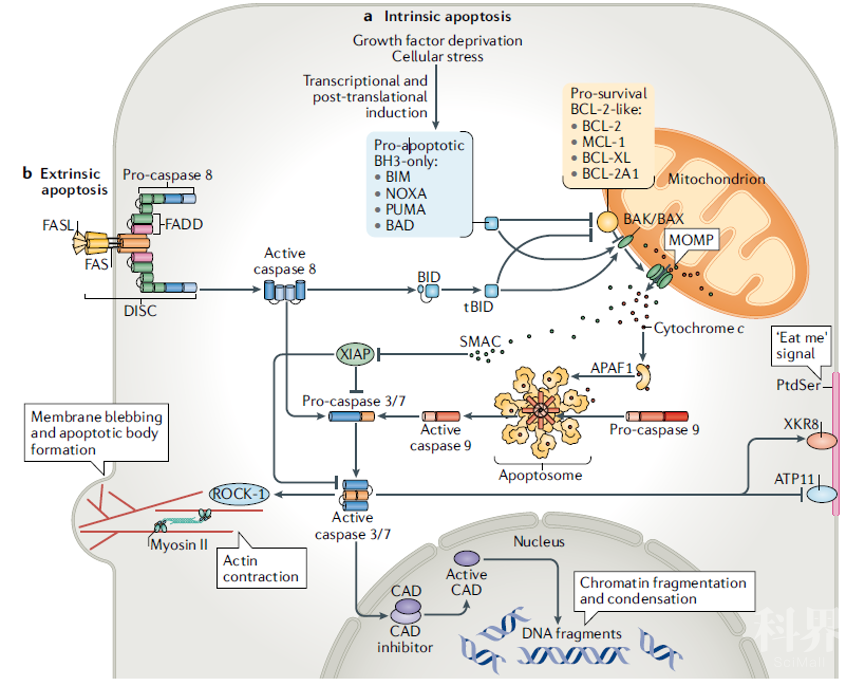
图2:凋亡途径激活的分子机制
内源性凋亡途径(a)由各种应激(例如,内质网应激,生长因子剥夺或DNA损伤)和发育信号激活。这在转录和/或翻译后诱导BCL-2家族的促凋亡成员,称为BH3家族蛋白,例如BIM,PUMA,BAD或NOXA。BH3-only蛋白结合并中和促存活BCL-2蛋白,如BCL-2,BCL-XL,MCL-1和BCL-2相关蛋白A1(BCL-2A1;BFL-1在人体内),从而释放细胞凋亡的关键效应物,BAK和BAX(BCL-2家族的第二个促凋亡亚组的成员),然后组装成大的复合物,导致线粒体外膜破裂(线粒体外膜透化或MOMP)。这导致凋亡因子的释放,例如细胞色素c和SMAC(Caspase-的第二线粒体激活剂,也称为DIABLO)。
据报道,某些BH3家族蛋白,例如BIM和PUMA,也直接结合并激活BAK和BAX以诱导MOMP。细胞色素c通过与凋亡肽酶激活因子1(APAF1)结合而引起凋亡小体的形成。启动子Caspase-9在该复合物中被激活,然后Caspase-9蛋白水解激活效应子Caspase-3和7。SMAC阻断Caspase-抑制剂XIAP的活性。
死亡受体诱导的细胞凋亡(b)由死亡受体的激活触发,例如存在于相邻细胞上的FAS配体(FASL)激活FAS。这导致通过衔接蛋白FADD(通过死亡结构域相关的FAS)将前Caspase-8募集到死亡受体的细胞内区域,导致形成所谓的死亡诱导信号复合物(DISC),其催化Caspase-8的活化。活性Caspase-8通过效应Caspase-3和7的直接蛋白水解活化或间接通过BH3-only蛋白BID蛋白水解活化成tBID诱导细胞杀伤,从而参与内在的凋亡途径。
通过任一途径激活的效应子Caspase-切割数百种细胞内蛋白质以诱导典型的凋亡形态并防止导致炎症反应的细胞内损伤相关分子模式(DAMP)的泄漏。这些效应器Caspase-直接或间接激活ROCK-1激酶,其通过肌动蛋白收缩和Caspase-活化的DNA酶(CAD)(通过切割其抑制剂ICAD)诱导质膜起泡,这导致核小体间DNA切割和染色质浓缩。效应器Caspase-还蛋白水解失活脂质翻转酶,例如ATP11,并蛋白水解激活脂质扰乱酶XKR8;总的来说,这导致磷脂酰丝氨酸(PtdSer)暴露于细胞膜的外部小叶上。这充当吞噬细胞的“吃我”信号并促进经历细胞凋亡的细胞的吞噬。
凋亡也可以通过死亡受体(也被称为外源性)途径,即死亡配体与细胞膜上的死亡受体(例如:TNF受体家族)结合(图2b)。细胞膜上的死亡受体与配体结合后活化,进而通过其细胞内结构域引起配体的聚集和下游Caspase-8的活化。活化的Caspase-8诱导下游caspase[22, 23]的激活。caspase 8的活性可以通过cFLIP (cellular CASP8 and FADD-like apoptosis regulator)调控[32]Caspase 8也可以将BID转化为其促凋亡形式tBID, tBID通过内在途径(见上文)触发BAK/ bax介导的MOMP和细胞凋亡[33]。
细胞焦亡
Caspase 1和Caspase 11(Caspase 4和Caspase 5是小鼠Caspase 11的人类同源物)在裂解型细胞死亡中具有重要作用,称为细胞焦亡,被广泛认为参与机体对病原菌的防御[34, 35]。在正常状态下Caspase 1被合成为无活性的酶原形式,其在称为炎症小体的复合体中自我催化活化(图3)。
炎症小体是在激活NLR(核苷酸结合结构域和富含亮氨酸重复序列)家族[36, 37]成员时形成的复合体,例如NLRP3(含有3的NLR家族pyrin结构域),通常通过激活衔接子来促进Caspase 1的活化,例如ASC(含有凋亡相关斑点样蛋白)NLRP3。NLRP3炎症小体可以被多种不同的信号激活,包括细胞内离子浓度的改变或未能降解可破坏溶酶体膜的惰性结构(如晶体)[37]。尽管已经发现众多的激活信号,NLRP3具体的激活机制尚不清楚。病原体衍生的DNA与AIM2的结合也促进炎症小体的组装。
炎症小体还可以结合NLRC4(含有CARD结构域),其在识别细菌组分的NAIP(NLR家族凋亡抑制蛋白)下游被激活,例如III型分泌(T3SS)装置的杆结构和鞭毛蛋白33。尽管炎症小体在其上游传感器中可能存在显着差异,因此它们的活化配体都会导致caspase 1活化。活性caspase 1能够切割多个下游靶蛋白,其中gasdermin家族成员gasdermin D(GSDMD)对细胞焦亡至关重要。GSDMD被Caspase-1切割成N-末端和C-末端片段(图3),后者在未剪切的GSDMD中发挥自身抑制活性。释放的N-末端GSDMD片段被募集到细胞膜的内部小叶中形成跨膜孔 (图3),导致钾流出和水流入,从而使质膜电位不稳定并导致细胞破裂。
除了激活GSDMD形成孔外,caspase 1还促进促炎细胞因子pro-IL-1β和pro-IL-18的前体蛋白水解转化为其生物活性形式,从而驱动广泛的炎症反应。这些细胞因子缺乏分泌所需的前导序列,细胞破裂使这些细胞因子释放到细胞外基质中,除了释放DAMPs38外,还提供炎症的进一步驱动因素。
近期研究结果表明,IL-1β和IL-18也可以通过GSDMD孔释放,即使只有少数这样的孔形成,并且当依赖于ESCRT机制的膜修复机制仍然存在时能够保持细胞活力39。然而,未来仍需要更多的研究来更好地确定这些事件发生的频率以及这种瞬时孔隙在细胞和生物体水平上对不同应激物的细胞反应的背景下的相关性。
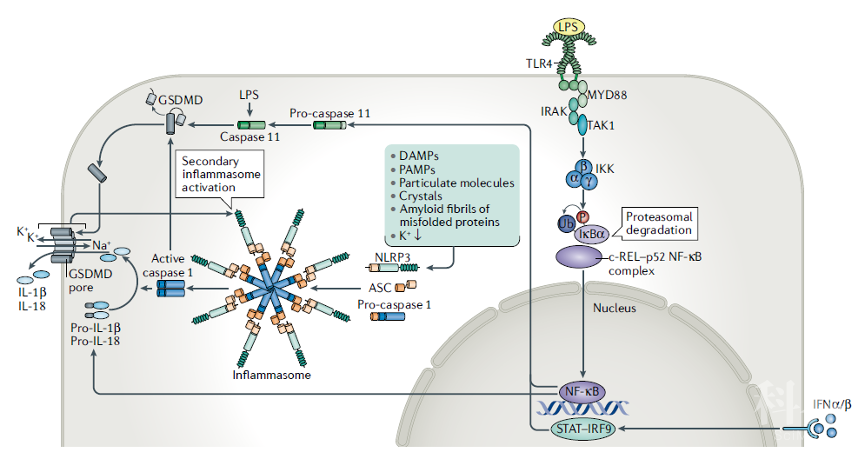
图3:炎症小体激活和细胞焦亡的分子特征。
遇到损伤相关分子模式(DAMP),病原体相关分子模式(PAMP),惰性结构或细胞内钾(K+)浓度紊乱的不同组合的细胞可经历炎症小体激活和随后的pyroptotic细胞死亡。在胞质溶胶中,上述刺激因素引起NLR家族受体的激活,例如NLRC4(含有NLR家族CARD结构域4)或NLRP3(含有NLR家族pyrin结构域3),触发了炎症小体的形成:作为Caspase-1活化位点的大蛋白质聚集体。
在细胞膜上,DAMP和PAMP可以触发Toll样受体,如TLR4,然后通过衔接蛋白MYD88激活IRAK1(IL-1受体相关激酶1)和丝裂原活化蛋白激酶TAK1。触发IKK(NF-κB激酶抑制剂)复合物磷酸化NF-κB途径抑制剂IκB,导致其遍在蛋白(Ub)依赖性蛋白酶体降解。这会释放c-REL–p52或RELA–p50 NF-κB复合物进入细胞核并激活依赖NF-κB的靶基因,例如编码pro-IL-1β,pro-IL-18和pro-caspase 11的靶基因。I型干扰素(例如IFNα/β)还可以通过激活细胞膜上的I型IFN受体并由此激活JAK-STAT途径信号传导来诱导前胱天蛋白酶11的表达。
活化的Caspase-1和11都可以切割gasdermin D(GSDMD)。GSDMD的N-末端片段通过孔形成引起质膜裂解。尽管Caspase-1在炎症小体中被激活,但已显示Caspase-11直接被细胞内脂多糖(LPS)激活。Caspase-1(但不是Caspase-11)也引起pro-IL-1β和pro-IL-18的蛋白水解转化为它们的生物活性形式,其通过GSDMD孔分泌或在细胞经历裂解时释放。ASC,凋亡相关斑点样蛋白;IRF9,干扰素调节转录因子9。
人Caspase-4和5及其鼠同源物Caspase-11在氨基酸序列上与Caspase-1高度同源。在小鼠中,Caspase-1和11的基因位置非常接近(这导致第一个Caspase-1敲除的动物也敲除Caspase-11)。
然而,Caspase-1和Caspase-11之间存在一些显着差异。尽管Caspase-11与Caspase-1相比具有类似的切割GSDMD效率,但Caspase-11不能将pro-IL-1β和pro-IL-18转化为它们的生物活性形式。Caspase 11也不同于Caspase 1,因为它不是组成型表达的,而是依赖于通过干扰素-α受体和/或NF-κB信号传导的转录诱导。
值得注意的是,与在模式识别受体下游激活的Caspase-1相反,Caspase-11及其人类直向同源物Caspase-4和5可以结合脂多糖(LPS)并被其直接激活。这表明Caspase-11在小鼠中以及人类中Caspase-4和5在宿主防御革兰氏阴性细菌中具有特别重要的作用。
事实上,caspase 11缺陷小鼠对LPS诱导的脓毒性休克具有抗性,表明LPS单独通过与Toll样受体TLR4结合引发的促炎反应不足以引起致死性脓毒性休克,但这需要LPS对caspase 11进行额外的细胞内活化。这些差异导致描述胱天蛋白酶11激活的机制涉及非经典炎症小体。破译caspase 1和caspase 11在体内的相对作用仍然具有挑战性,因为通过激活GSDMD的caspase 11的孔诱导能力促进了继发性NLRP3炎症小体激活。这很可能是由由此产生的钾外流驱动的,从而基本上将Caspase-11“重新连接”到所有的促炎信号,包括Caspase-1介导的生物活性IL-1β和IL-18的产生。
细胞坏死性凋亡
坏死性凋亡是程序性裂解细胞死亡的途径,其被认为在某些退行性或炎性疾病期间在杀死病原体感染的细胞和/或受损细胞中起作用。坏死性凋亡可以通过多种先天免疫信号传导途径诱导,包括通过刺激RIG-I样受体,TLR和死亡受体引发的途径。这些信号通路都导致坏死性激酶RIPK3(受体相互作用的丝氨酸-苏氨酸激酶3)的磷酸化和活化(图4),在死亡受体诱导的坏死性凋亡的情况下也需要RIPK1活性。RIPK3通过磷酸化激活假激酶MLKL(混合谱系激酶结构域样),引起其构象变化和活化,活化的 MLKL定位到质膜引起膜通透性变化 。
尽管由此导致的膜完整性和细胞溶质渗透压的丧失解释了与坏死性凋亡相关的细胞死亡的溶解特征,但是需要更多的工作来确定MLKL是否确实形成明确的孔,如GSDMD的情况,或导致以其他方式破坏质膜。与细胞焦亡一样,在MLKL激活的早期阶段,涉及ESCRT的细胞膜修复过程似乎能够停止甚至可能阻止细胞杀伤。

图4:诱导坏死性凋亡。
当凋亡抑制剂cIAP1和cIAP2被抑制并且Caspase-8活性被阻断时(参见图6),肿瘤坏死因子(TNF)与肿瘤坏死因子受体1(TNFR1)的结合导致RIPK1(受体相互作用的丝氨酸-苏氨酸激酶1)通过衔接蛋白TRADD(通过死亡域相关的TNFRSF1A)募集和磷酸化。RIPK1反过来磷酸化并由此激活RIPK3。RIPK3也可以通过涉及衔接子TRIF的过程通过Toll样受体(TLR)信号传导来激活。活性RIPK3磷酸化MLKL。这导致MLKL的构象变化,使其易位并破坏质膜的完整性(可能通过形成孔)。由此产生的水和钠(Na+)流入,钾流出,导致细胞肿胀,膜电位破坏,最终细胞裂解。具有细胞死亡调节剂cFLIP(细胞CASP8和FADD样凋亡调节剂)的长同种型的Caspase-8的异二聚体可以切割RIPK1,从而抑制TNFR1介导的坏死性凋亡的诱导。
自噬性细胞死亡
一种独特的程序性细胞死亡(PCD)途径,不需要caspases,并且不具有细胞凋亡,坏死性凋亡或细胞凋亡的典型形态学特征。在体内,这种死亡方式对于黑腹果蝇发育过程中唾液腺的退化非常重要,此外与培养的哺乳动物细胞的死亡,和细胞凋亡缺陷的哺乳动物细胞在营养缺乏条件下的死亡。自噬细胞死亡的一个典型特征是在死亡细胞的胞浆内积累大的液泡[11]。
氧中毒死亡
氧中毒死亡是一种不依赖于Caspases的,非炎性,凋亡样细胞死亡途径,一般在病毒感染时引起大量活性氧(ROS)释放的情况下诱导发生。据报道需要ROS传感器KEAP1,磷酸酶PGAM5和促凋亡因子AIFM1 [12]。
铁死亡
依赖于铁的、非凋亡性氧化的细胞死亡形式,在形态、生化和分子上都不同于细胞凋亡、坏死、焦亡和自噬。它类似于谷氨酸诱导的细胞死亡,阻断细胞半胱氨酸摄取,干扰抗氧化细胞防御,最终导致细胞死亡[13]。铁死亡是由细胞依赖谷胱甘肽的抗氧化防御失效或阻断引起的。这导致不受抑制的脂质过氧化,并最终杀死细胞。亲脂性抗氧化剂和铁螯合剂可以防止铁腐蚀细胞的死亡。
继发性坏死
继发性坏死是一个非程序性的继发事件,发生在细胞凋亡的细胞中,当它们在早期没有被吞噬细胞吞噬和消化时-例如,由于对广泛细胞凋亡的吞噬能力超负荷,在高效抗癌治疗后可能发生,其在短时间内引起大量恶性细胞的凋亡。经历继发性坏死的细胞的形态使人联想到非程序性的坏死性死亡,例如由细胞燃烧或冷冻引起的坏死性死亡。
NETosis
NETosis是活化的嗜中性粒细胞杀死入侵病原体的机制之一。在这个过程中,嗜中性粒细胞释放颗粒蛋白和染色质,形成称为嗜中性粒细胞胞外陷阱(NETs)的细胞外纤维以捕获和杀死病原体。由于重要细胞物质的释放,NETosis通常与中性粒细胞死亡有关,并且在形态学上不同于细胞凋亡或坏死 [14]。
CTL和NK细胞诱导的细胞杀伤
病毒感染的细胞或恶性细胞可以被活化的CD8+细胞毒性T淋巴细胞(CTL)和自然杀伤(NK)细胞以具有裂解和非裂解PCD特征的方式杀死[15]。由CTL和NK细胞分泌的穿孔素在其靶细胞的质膜中诱导孔(类似于补体系统)并允许颗粒酶进入细胞的胞质溶胶[16]。由此导致的细胞完整性丧失导致细胞死亡。据报道,颗粒酶通过多种机制杀死靶细胞,包括通过蛋白水解激活BH3-only蛋白BID[17]或蛋白水解激活效应caspases诱导细胞凋亡,以及通过蛋白水解诱导细胞凋亡样细胞死亡激活gasdermin E [18]。活化的CTL和NK细胞也可以使用膜结合的FAS配体或TNF触发死亡受体诱导的细胞凋亡。IFNγ在介导T细胞和NK细胞的细胞毒性作用中也起重要作用。体外研究表明,IFNγ可以通过上调BH3-only蛋白BIM来激活内在的凋亡途径,并且还可以通过增加TRAIL和TNFR1的表达来增强死亡受体诱导的细胞凋亡 [19, 20]。
来源:BioGossip BioArt
原文链接:http://mp.weixin.qq.com/s?__biz=MzA3MzQyNjY1MQ==&mid=2652514009&idx=3&sn=cf2822f16e35f93980af9e037d92cacb
版权声明:除非特别注明,本站所载内容来源于互联网、微信公众号等公开渠道,不代表本站观点,仅供参考、交流、公益传播之目的。转载的稿件版权归原作者或机构所有,如有侵权,请联系删除。
电话:(010)86409582
邮箱:kejie@scimall.org.cn