来源:BioArt
肠易激综合征(Irritable Bowel Syndrome,IBS)是一种全球流行的肠道功能紊乱性疾病,以反复出现腹痛、排便习惯和(或)大便性状改变为主要临床表现,IBS主要见于女性。基于主要的大便性状将IBS分为便秘型(IBS-C),腹泻型(IBS-D)或混合(IBS-M)型。IBS发病机制涉及胃肠动力的变化,肠道分泌物,内脏超敏反应和肠道渗透性,研究表明所有这些都与肠道微生物有关。但是,由于动物研究和人类研究之间存在明显的脱节,同时缺乏针对疾病特异性生理变化的综合多组学观点,因此很难确定肠道微生物在IBS中的作用机理。
2020年9月10日,美国梅奥诊所消化内科和肝病科Purna C. Kashyap研究团队和明尼苏达大学生物科学学院Dan Knights团队合作在Cell上发表了题为 Longitudinal Multi-omics Reveals Subset-Specific Mechanisms Underlying Irritable Bowel Syndrome 的文章,研究团队对IBS宿主生理进行了多组学测量的纵向研究(包括肠道微生物组,代谢组,宿主表观基因组和转录组),最终确定了IBS亚类型特异性、症状相关的微生物组成和功能变异,其中一组已确定的微生物代谢产物变异子集与IBS有关的宿主生理机制相关。通过整合多个数据层,研究团队将嘌呤代谢确定为一种新型IBS宿主-微生物代谢途径,同时嘌呤饥饿被确认为潜在的IBS治疗靶标(图1)。

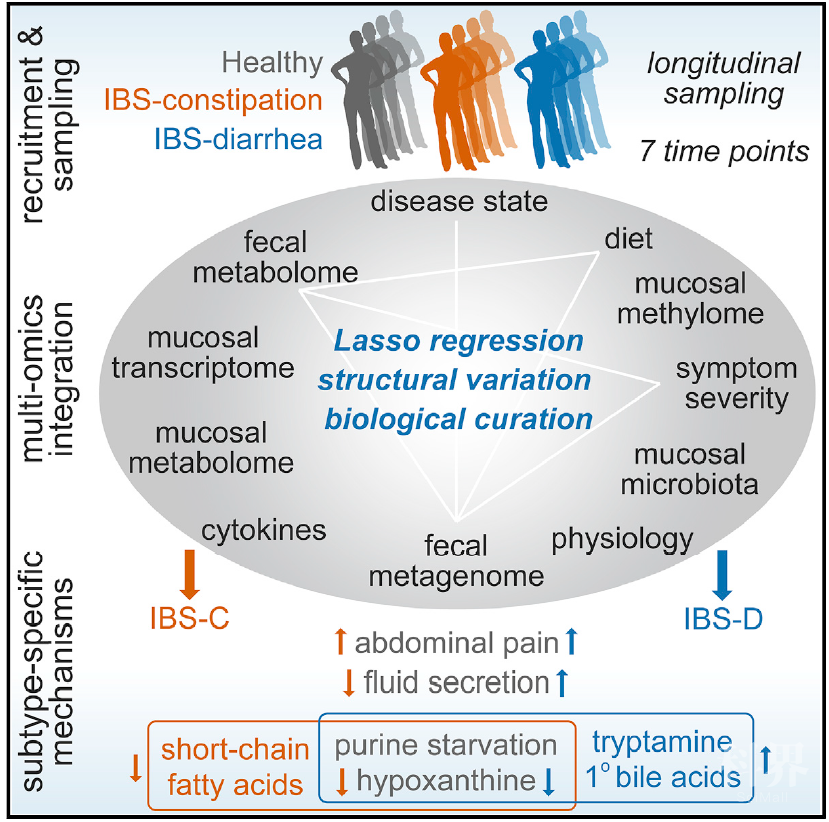
图1.图形概要
以往针对慢性胃肠道疾病中肠道微生物组研究主要采用横断面研究,该研究提供了高度动态的生态系统的快照,但是横断面研究存在很大的缺陷:无法解决异质性难题。研究团队独辟蹊径,采用了纵向采样对肠道微生物组成分变化进行检测。所谓纵向采样研究即在不同时间点对健康志愿者和IBS患者进行样本采集并进行多组学分析,然后对纵向数据进行二次抽样,测试重要的分类单元,以及将单个时间点的结果与所有时间点上受试者平均结果进行比较。研究团队发现纵向采样能够识别出横断面研究可能遗漏的微生物变化。
纵向采样研究揭示随着时间流逝,相对健康志愿者(HC)和IBS-D患者,IBS-C微生物群的组成表现出更大变异性。此外,IBS症状严重程度与肠道菌群的功能变异相关。利用KEGG Orthology (KO) term在粪便宏基因组学数据中的丰度研究微生物功能变异,结果发现,有74个KO term与严重IBS-C患者相关,44个KO term与严重IBS-D的患者相关(FDR of <0.1)。与轻度、中度IBS相比,重度IBS-C和IBS-D中均发现了醇脱氢酶(ADH酶)的KO term。此外,这些数据表明ADH活性可能与腹部疼痛相关。以上结果表明,IBS病理生理(粪便性状记录和排便前腹痛)与特定的细菌和代谢产物有关。
那么肠道菌群驱动IBS病理生理症状的具体机制是什么呢?作者结合代谢组学(量化微生物组的代谢输出,即中腔和粘膜相关样品的生化反应事件)与生理学进行了深入的研究。代谢组学结果发现,与HC相比,IBS-C患者的粪便样本中短链脂肪酸(SCFA),如丙酸盐、丁酸盐和乙酸盐都明显降低。作者还发现色氨酸和色胺在IBS-D患者的粪便样本中显着增加,这可能部分导致了IBS-D患者粪便中增加的水分含量,因为作者之前的研究发现细菌衍生的单胺色胺可激活血清素4导致悉生小鼠体内体液分泌增加和转运时间减少。此外,由于某些形式的胆汁酸(BA,例如羟基化BA—鹅去氧胆酸)被发现具有增加人类的肠液分泌的功能,作者的研究结果发现了与IBS相关的BA属性差异:与HC相比,粪便样本中未结合的初级BA量在IBS-D患者中明显更高,而在IBS-C患者显着降低。以上表明,IBS-D患者粪便中较高的水含量与粪便中色胺和原发性BA的增加有关。数据整合结果证明,肠道微生物代谢产物的改变是IBS症状加重的微生物基础。这也进一步强调了多组学数据整合的重要性。
那么,是哪些微生物代谢途径参与了IBS病理生理变化呢?作者利用非针对性代谢组学方法来鉴定推动IBS病理生理变化的潜在新型微生物途径,结果发现,与HC的比较,在IBS-C患者的粪便样本中,赖氨酸、尿嘧啶和次黄嘌呤均明显降低。随后,作者通过宏基因组学功能分析发现,相对于HC,IBS-C患者的黄嘌呤脱氢酶/氧化酶(XO)和黄嘌呤磷酸核糖基转移酶的KO terms模块水平升高,提示IBS患者中肠道菌群可能促进嘌呤的消耗。对次黄嘌呤代谢更深入的宏基因组KO terms分析发现,IBS患者的次黄嘌呤利用和分解增强,这也与IBS-C患者粪便中较低的次黄嘌呤相一致。这表明嘌呤代谢途径参与了IBS病理生理。
为了进一步阐明微生物对IBS中不同代谢产物丰度的贡献,作者利用最近发现的将结构可变的基因组区域与代谢产物丰度相关联的方法(SV关联分析),该分析方法可以识别涉及生产或消耗代谢产物的微生物基因,主要是通过识别微生物的缺失区(DR)或可变区域(VR)来确定某些微生物。作者观察到的最强VRs关联来自于Lachnospiraceae sp. 3_1_46FAA菌的一个单区域与次黄嘌呤的关联。作者随后选择了与Lachnospiraceae sp. 3_1_46FAA菌基因组相似的2种Lachnospiraceae菌株,发现这2种菌的生长培养基中次黄嘌呤明显减少。而移植了Lachnospiraceae菌的无菌小鼠的盲肠内次黄嘌呤水平较低。这进一步表明肠道微生物参与了IBS肠道嘌呤代谢途径的调控。
由于次黄嘌呤是一种宿主-微生物共代谢物,提示次黄嘌呤的多少受微生物和宿主代谢的共同影响。为了确定宿主对次黄嘌呤库减少的贡献,作者检查了患者的结肠活检组织中嘌呤代谢基因的表达变化。人的黄嘌呤氧化酶(由XDH基因编码)的基因在IBS-C和IBS-D患者的结肠活检样本中均表达升高。这表明,微生物组和宿主的黄嘌呤氧化酶活性共同参与了次黄嘌呤库的耗尽。肠上皮细胞从头合成嘌呤的能力有限,因而主要依靠腺苷酸生物合成过程中的嘌呤再生途径(图2)。因此,作者检查了嘌呤再生途径中潜在的转录变化。结果发现,IBS-C和IBS-D患者的嘌呤核苷磷酸化酶(PNP,嘌呤再生途径中的第一个基因)的表达比HC中高约2倍。而IBS患者PNP表达的上调与次黄嘌呤水平之间存在负相关。以上结果表明,微生物组和宿主共同参与了次黄嘌呤库的耗尽。
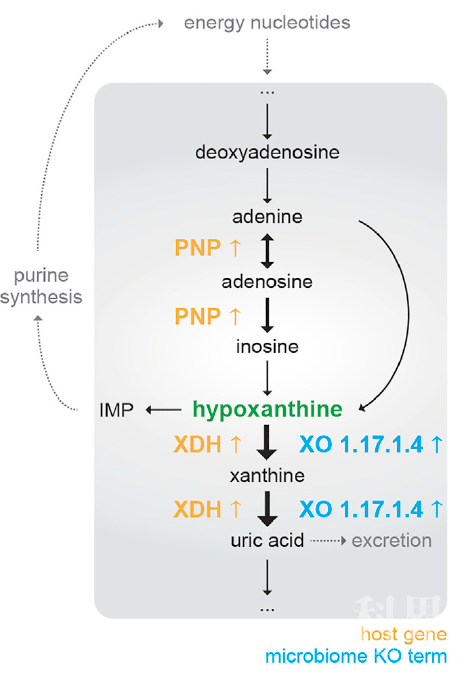
图2. 人-微生物组嘌呤核苷酸降解途径中IBS相关变化
总的来说,研究团队发现并提出了一种模型:微生物和宿主共同参与了嘌呤核苷酸的消耗,从而诱导了结肠组织的代谢压力。反过来,这可能导致一种通过增加嘌呤再生诱导的补偿反应(图2)。使用多组学视角,研究团队认为低水平的嘌呤核苷酸可能导致黏膜修复过程中较低的上皮能量状态,这可能部分解释IBS的病理生理学。该研究结果强调了纵向采样和整合互补的多组学数据在研究IBS病理生理机制中的重要性,该机制的研究将为慢性胃肠道疾病的综合治疗策略提供治疗靶标。
来源:BioGossip BioArt
原文链接:https://mp.weixin.qq.com/s?__biz=MzA3MzQyNjY1MQ==&mid=2652499138&idx=4&sn=cd74640a40459a6e76ea6da900f7b6f5&chksm=84e27f76b395f66055e07de406d64250e2426c6167e6dd08216e0717f48d35f2526170498501#rd
版权声明:除非特别注明,本站所载内容来源于互联网、微信公众号等公开渠道,不代表本站观点,仅供参考、交流、公益传播之目的。转载的稿件版权归原作者或机构所有,如有侵权,请联系删除。
电话:(010)86409582
邮箱:kejie@scimall.org.cn



