来源:逻辑神经科学
小胶质细胞(microglia)功能失调与神经退行性变密切相关,其中就包括阿尔茨海默症(AD)的发病机制,然而调控致病性小胶质细胞的基因表达的机制尚不清楚。
研究发现,绝大多数AD进展的常见风险因子,或者特异性地在小胶质细胞表达,亦或者是小胶质细胞中的表达明显高于大脑的其他细胞【1】。此外,小胶质细胞增生是许多神经疾病的一种常见组织学特征;而且由小胶质细胞和星形胶质细胞所调控的慢性神经炎症是AD的病理学特征之一。
研究也表明,转录因子CCAAT/增强子结合蛋白β(CCAAT/enhancer binding protein beta,简称c/EBPβ)调节小胶质细胞的促炎基因【2】;而且c/EBPβ的蛋白水平在AD中上调【3】。c/EBPβ调控小胶质细胞的转录程序,促进炎症和神经元细胞死亡。
基于以上研究背景,美国基因泰克(Genentech)公司的Vishva M. Dixit博士和Kim Newton博士团队旨在揭示该转录因子在小胶质细胞内中是如何被调控的。其相关研究已于2020年8月13日以Ubiquitin Ligase COP1 Suppresses Neuroinflammation by Degrading c/EBPβ in Microglia为题在线发表于Cell。 首先,值得注意的是,研究团队发现,在小鼠骨髓源性巨噬细胞中(BMDMs)中,c/EBPβ的翻译后机制受到严格的调控;但这并不能反映在其转录本中的变化。所以,紧接着,研究者通过质谱的方法在BMDMs中鉴定了一种c/EBPβ的负调节因子COP1,也称RFWD2,是一种多亚基cullin-RING泛素连接酶CRL4COP1/DET1的底物接合分子(substrate adaptor)。同时,团队也发现:COP1对于小鼠原代小胶质细胞和巨噬细胞中c/EBPβ蛋白水平的翻译后抑制至关重要。
首先,值得注意的是,研究团队发现,在小鼠骨髓源性巨噬细胞中(BMDMs)中,c/EBPβ的翻译后机制受到严格的调控;但这并不能反映在其转录本中的变化。所以,紧接着,研究者通过质谱的方法在BMDMs中鉴定了一种c/EBPβ的负调节因子COP1,也称RFWD2,是一种多亚基cullin-RING泛素连接酶CRL4COP1/DET1的底物接合分子(substrate adaptor)。同时,团队也发现:COP1对于小鼠原代小胶质细胞和巨噬细胞中c/EBPβ蛋白水平的翻译后抑制至关重要。接下来,染色质免疫沉淀反应测序(ChIP-seq)分析显示,在原代小胶质细胞和BMDMs中,COP1的敲除会导致c/EBPβ与远端增强子元件(组蛋白H3上赖氨酸4的三甲基化(H3K4me3)、组蛋白H3上赖氨酸27的乙酰化(H3K27ac))的结合;在对c/EBP远端结合位点的从头基序分析(De novo motif analysis)中,发现c/EBPβ通过与促炎基因增强子域的结合来调控促炎基因的表达。已有研究表明,小胶质细胞增强子的遗传变异与AD的发病机制有关【4】,同时,实验也显示,当在小胶质细胞中敲除COP1,神经元的死亡明显增多。这部分结果则说明:c/EBPβ主要通过增强子区域来调控促炎基因的表达。
那么,再接下来的另一个问题是:COP1敲除的小胶质细胞是否对c/EBPβ所导致的神经毒性更加易感呢?结果首先证实,在小胶质细胞中敲除COP1会促使c/EBPβ的迅速积累、并驱动强有力的促炎和神经退化相关基因的表达。而在敲除COP1的小胶质细胞中,再敲除Cebpb(编码c/EBPβ),神经毒性得到了阻止,且趋化因子和细胞因子的分泌也降低了,即缓解了神经炎症,增加了神经元存活。这部分结果提示:COP1敲除小胶质细胞在体外表现出c/EBPβ依赖性的神经元杀伤。
激活性小胶质细胞可以通过补体级联通路直接攻击神经元,这一个过程由C1q和经典途径所启动【5】。结果进一步显示,由敲除COP1小胶质细胞所引发的神经元丢失可以被促炎性细胞因子的抗体明显阻止,特别是C1q抗体,该抗体可以在神经元中阻断C1q的结合。因此,作者认为:致使神经元死亡的神经毒性由补体的激活所导致。
研究最后,值得关注的是,补体通路的过度激活与各种神经系统疾病中的神经元损伤有关,其中,C3蛋白尤其在伴有tau病理的AD人脑突触中进行积累【5】;而且AD患者脑脊液中完整的和加工过的C3水平均升高,并与tau蛋白水平相关【6】。所以,小胶质细胞中Cop1的缺失是否会影响表达人源突变Tau蛋白(P301S)的转基因小鼠的AD样疾病进展? 的确,实验证实:c/EBPβ促进COP1敲除小胶质细胞中C3基因的表达,这一结果与之前的报道相一致:c/EBPβ是C3基因表达的主要转录因子【8】。然后的实验也表明,较未敲除COP1的Tau-P301S小鼠而言,敲除COP1小鼠的神经退行性变和海马萎缩更加明显,且脑萎缩程度与异常磷酸化Tau水平上升、与小胶质细胞增生和星形胶质细胞增生的加重相一致。这些实验则综合表明:Cop1的缺失加速了Tau介导的AD小鼠模型疾病进展,激活性的小胶质细胞发挥了有害的作用。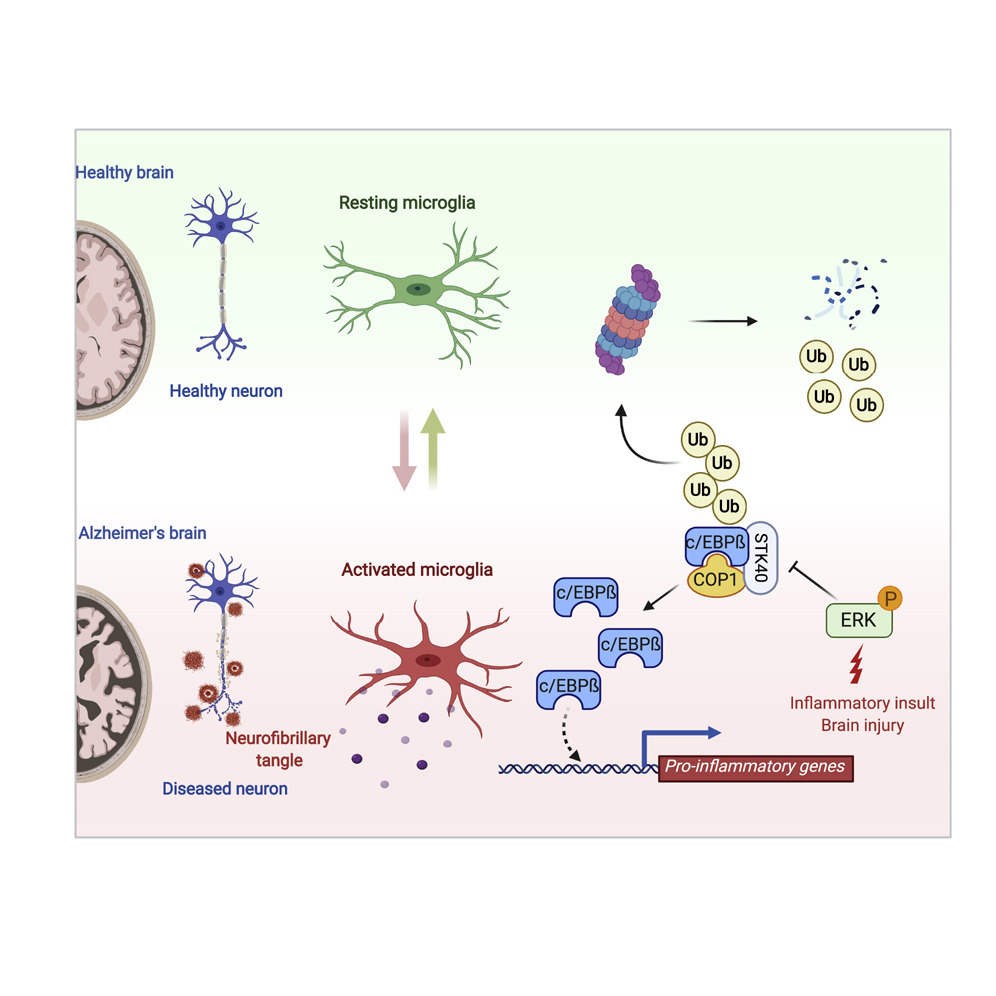
文章结论这项研究确定COP1可以作为由c/EBPβ介导的神经炎症的“刹车”。在激活性刺激(如脂多糖)条件下,COP1以一种依赖于ERK(即细胞外信号调节激酶)的方式迅速失活,这致使c/EBPβ的快速积累和促炎基因的表达。而该“刹车”的缺失可能导致慢性神经炎症,而神经炎症会促进AD的发病机制。
该研究也提示,COP1是小胶质细胞中致病性c/EBPβ依赖基因表达的重要抑制因子。
最后,c/EBPβ杂合性所赋予的细胞保护作用提示了该转录因子可能是一个药物靶点。虽然,目前研究者认为转录因子是很难被靶向的,但是降解型的化学诱导剂可能是靶向c/EBPβ的一种选择。
原文链接:
https://doi.org/10.1016/j.cell.2020.07.011
参考文献(上下滑动查看)
【1】Lambert, J.C., et al.. European Alzheimer’s Disease Initiative (EADI); Genetic and Environmental Risk in Alzheimer’s Disease; Alzheimer’s Disease Genetic Consortium; Cohorts for Heart and Aging Research in Genomic Epidemiology (2013). Meta-analysis of 74,046 individuals identifies 11 new susceptibility loci for Alzheimer’s disease. Nat. Genet. 45, 1452–1458.
【2】Straccia, M., et al.. (2011). Pro-inflammatory gene expression and neurotoxic effects of activated microglia are attenuated by absence of CCAAT/enhancer binding protein b. J. Neuroinflammation 8, 156.
【3】Wang, Z.H., et al. (2018). C/EBPb regulates deltasecretase expression and mediates pathogenesis in mouse models of Alzheimer’s disease. Nat. Commun. 9, 1784.
【4】Nott, A., et al. (2019). Brain cell type-specific enhancerpromoter interactome maps and disease-risk association. Science 366, 1134–1139
【5】Dejanovic, B., et al. (2018). Changes in the Synaptic Proteome in Tauopathy and Rescue of Tau-Induced Synapse Loss by C1q Antibodies. Neuron 100, 1322–1336.
【6】Kirk, R.A., Kesner, R.P., Wang, L.M., Wu, Q., Towner, R.A., Hoffman, J.M., and Morton, K.A. (2019). Lipopolysaccharide exposure in a rat sepsis model results in hippocampal amyloid-b plaque and phosphorylated tau deposition and corresponding behavioral deficits. Geroscience 41, 467–481.
【7】Hernandez-Encinas, E., et al.. (2015). CCAAT/enhancer binding protein b directly regulates the expression of the complement component 3 gene in neural cells: implications for the pro-inflammatory effects of this transcription factor. J. Neuroinflammation 12, 14.
来源:LT-Neuroscience 逻辑神经科学
原文链接:https://mp.weixin.qq.com/s?__biz=MzI4Mjk3NzUxOQ==&mid=2247487332&idx=1&sn=b3f1bcd9e0c3636474a6528c693a5fee&chksm=eb90fae4dce773f2bdfff4343328e2b31eceddd9cbb945fcf525a171ae0f884dda6339bb48ef#rd
版权声明:除非特别注明,本站所载内容来源于互联网、微信公众号等公开渠道,不代表本站观点,仅供参考、交流、公益传播之目的。转载的稿件版权归原作者或机构所有,如有侵权,请联系删除。
电话:(010)86409582
邮箱:kejie@scimall.org.cn



