来源:BioArt
撰文 | 咸姐
责编 | 兮
心肌肥大是心肌细胞对各种外在和内在刺激的反应,这些刺激会增加生物力学的压力。虽然肥厚最终能使肠壁张力正常化,但它仍能导致不利的结果,如猝死或心力衰竭【1】。研究发现,氧化应激是引起心肌肥厚尤其是肾上腺素能性肥大的一个重要原因。氧化应激是一种病理学上高水平的细胞内氧化剂,可能会推动心血管疾病的进展,这一概念已经引发了许多干预措施的临床试验,如抗氧化维生素。但这并没有降低疾病的发展和风险,部分原因可能是人们关注氧化应激的危害,却忽视了氧化还原平衡、活性氧(ROS)和活性氮在细胞生理病理中的广泛作用【2】。
氧化还原信号——一种对细胞内信号通路组分进行特定的、通常可逆的氧化/还原修饰的活性反应组分——在许多生理和病理过程中被认为是至关重要的【3】。而其中一个很重要的修饰就是8-氧代鸟嘌呤(8-oxoguanine,o8G)。核酸中鸟嘌呤氧化产生o8G,可以与腺嘌呤配对,诱导DNA中鸟嘌呤向胸腺嘧啶(G>T)的突变【4】,而研究发现在各种疾病发病过程中, RNA比DNA更容易产生o8G修饰。
在心脏中,氧化还原信号可以调节多种生理过程,并参与多种病理生理过程和内稳态或应激反应途径。miRNA作为靶向mRNA的转录后调节因子,其氧化已被发现与细胞的氧化还原状态相关,同时多种miRNA被证实参与了心肌肥厚发病过程。但是,迄今为止,o8G氧化修饰仅被认为是氧化损伤的结果,而其与miRNA相关的有效的表观转录水平作用从未被提及,并且其是否与心肌肥大有关也未可知。
2020年8月5日,来自韩国高丽大学的Sung Wook Chi研究团队在Nature上在线发表文章“Position-specific oxidation of miR-1 encodes cardiac hypertrophy”,在氧化还原相关的心肌肥厚动物和细胞模型中对氧化的miRNA进行了特异性测序,发现miR-1种子区域可以产生位置特异性o8G修饰,并通过o8G·A碱基配对调控新的mRNA,从而诱导心肌肥厚的发生。由此表明miRNA的位置特异性氧化可以作为一种表观转录机制来协调病理生理氧化还原所介导的基因表达。

首先,体外细胞实验和体内小鼠模型均证实,肾上腺素能受体抑制剂(苯肾上腺素PE或异丙肾上腺素ISO)可刺激心肌肥厚的发生,促使细胞产生ROS。此外,血清饥饿(刺激肾上腺素能性心肌肥厚的前提条件)可以增强高基础ROS水平下的心肌增大表型。而抗氧化剂N-乙酰半胱氨酸(NAC)则可以逆转这些现象。同时,研究发现在PE处理过的大鼠心肌细胞(H9c2细胞)及原代大鼠心肌细胞(rCMC)中,小RNA尤其是20 nt大小左右的miRNA中出现了o8G修饰的富集,这比细胞中出现在大RNA中的o8G的程度更高,也远远高于由百草枯诱导的氧化应激所出现的o8G修饰的数量。
随后,本文研究人员通过优化免疫沉淀技术和o8G诱导的G>T转换,开发了一种针对miRNA中o8G的测序方法,即o8G-miSeq。将该方法应用于H9c2细胞或rCMC中,结果均显示,miR-1b比其他miRNA更易被氧化,o8G富集程度更高,并且在PE处理后,o8G主要发生在种子区域,其中第2、3、7和15位置的o8G明显增加(图1 左)。而当对PE处理的rCMC进行血清饥饿时,miR-1b的第7位碱基G成为唯一被氧化的碱基(图1 右),miRNA测序分析表明仅在这个位置G>T转换增加了约2倍。

图1 o8G-miSeq分析PE处理后的rCMC(右:进行血清饥饿)
进一步的实验证实,虽然在ISO处理的小鼠心脏中,miR-1的表达降低,但是其氧化水平是增强的。那么发生在其种子区域的o8G(位置2、3、7,分别命名为2o8G-miR-1、3o8G-miR-1 和7o8G-miR-1)是否可使得miR-1通过o8G·A碱基配对而识别新的靶点呢?结果是肯定的。荧光素酶报告实验显示o8G-miR-1可以通过同源的含氧位点使靶基因沉默,这些含氧位点可以对PE处理产生反应,并在NAC处理后反应减弱,从而表明内源性o8G-miR-1的水平足以重新靶向目的基因并实现基因沉默。当然,由于残存的o8G·C碱基配对活性,氧化的miR-1也能够抑制与种子序列匹配的一些靶基因,只是抑制效果远远不如未被氧化的miR-1。
已知miR-1可以诱导细胞萎缩,但是本文研究人员发现PE处理可以损伤miR-1的这种作用。当向心肌细胞中导入合成的2o8G-miR-1、3o8G-miR-1 和7o8G-miR-1时,细胞表现出比PE处理后更严重的肥厚表型,当用碱基U替代o8G(形成2U-miR-1、3U-miR-1和7U-miR-1)时也出现了同样的表型,由此表明o8G对心肌肥厚的诱导是依赖于其o8G·A碱基配对活性的。进一步的,研究发现,由7o8G-miR-1或7U-miR-1诱导产生的心肌肥厚,其细胞内心房肽ANP(一种心肌肥厚的标志物)的表达显著增加,随着时间的增加,细胞逐渐增大。将合成的7o8G-miR-1或7U-miR-1通过尾静脉注射给予小鼠后,小鼠心脏室间隔心肌细胞增大约19%,心脏增大至少10%(图2)。同样的,心肌细胞特异性表达7U-miR-1的转基因小鼠也表现出类似的表型。而当特异性抑制小鼠心肌细胞中的7o8G-miR-1时,ISO诱导的心肌肥厚现象减弱。
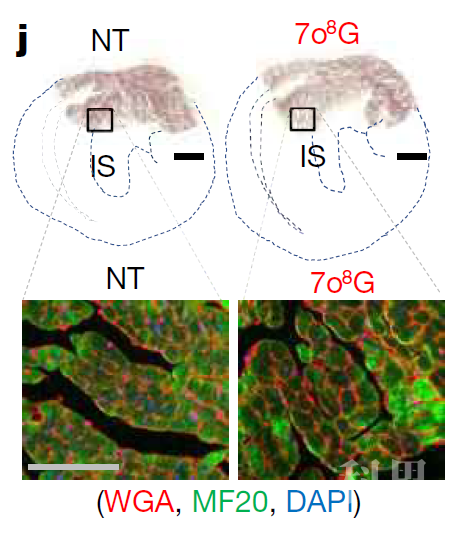
图2 7o8G-miR-1给药后小鼠心脏组织切片
对o8G-miR-1给药后的心肌细胞进行RNA测序以评估miR-1的氧化带来的转录组效应,研究人员发现miR-1的o8G氧化修饰使得其重新靶向了一批新的mRNA,miR-1种子区域的氧代位点(如7氧代位点)在3’-UTR区域进化保守,使大多数基因表达下调,而这些表达下调的差异基因功能主要与心脏功能如心脏形态发生、心脏收缩和钙离子转运相关,并富集于心肌肥厚相关通路中。
最后,研究人员发现,o8G-miR-1也参与了其他类型心肌病的发生,在心肌病病人的心肌细胞中,miR-1种子区域的氧代位点靶向的mRNA也出现富集,例如HSPB7——先天性扩张性心肌病的风险基因——具有7氧代位点。
综上所述,本文证明了肾上腺素能受体的激活产生ROS,导致miR-1种子区域o8G的形成(尤其是形成7o8G-miR-1),从而使得miR-1重新靶向抑制可在心肌肥厚通路中发挥作用的新靶点,进而驱动心肌肥厚和疾病的发生。也由此表明,o8G在miRNA中的形成可能是一种普遍的表观转录机制,通过这种机制,ROS在病理生理条件下可以对基因表达进行微调,作为氧化还原信号的一部分,最终导致表型的改变。
来源:BioGossip BioArt
原文链接:https://mp.weixin.qq.com/s?__biz=MzA3MzQyNjY1MQ==&mid=2652494840&idx=6&sn=eab3fba10c0c490b6790b1b412eac28a&chksm=84e2484cb395c15ade7ca253d810bc81f5d1179bdd8a02eb3e277d3815a0f264c61aca9f4308#rd
版权声明:除非特别注明,本站所载内容来源于互联网、微信公众号等公开渠道,不代表本站观点,仅供参考、交流、公益传播之目的。转载的稿件版权归原作者或机构所有,如有侵权,请联系删除。
电话:(010)86409582
邮箱:kejie@scimall.org.cn

