来源:宏基因组
正文分析细菌群落的最常见方法是对保守的16S rRNA基因进行测序。由于菌株的变异,不能使用16S rRNA基因序列数据直接鉴定功能概况,因此已经开发了几种方法来单独根据分类学概况(扩增子序列)预测微生物群落功能。随机宏基因组测序(Shotgun metagenomics sequencing, MGS)对整个基因组而不是标记基因进行测序,也可用于表征群落的功能,但是如果在活检样本中或存在群落生物量很少时容易存在宿主污染。
PICRUSt(以下简称“ PICRUSt1”)是为从16S标记序列预测功能而开发的,已被广泛使用,但有一定的局限性。标准PICRUSt1工作流程要求输入序列是根据Greengenes数据库的兼容版本进行有参比对而生成的OTU表。由于对参考OTU的这种限制,默认PICRUSt1工作流程与序列去噪方法不兼容,后者会产生扩增子序列变体(ASV)而不是OTU。ASV具有更好的分辨率,可以更容易地区分密切相关的生物。此外,PICRUSt1使用的细菌参考数据库自2013年以来未进行更新,并且缺少成千上万个最近添加的基因家族。
我们期望优化基因组预测将提高功能预测的准确性。因此,PICRUSt2算法(图1a)包括优化基因组预测的步骤,包括将序列置于参考系统发育树中,而不是限于有参OTU的预测(图1b);基于更大的参考基因组和基因家族数据库的预测(图1c);更严格地预测途径丰度(补充图1);并能够预测复杂的表型和集成自定义数据库。
图1:PICRUSt2 算法原理示意图Fig. 1: PICRUSt2 algorithm.
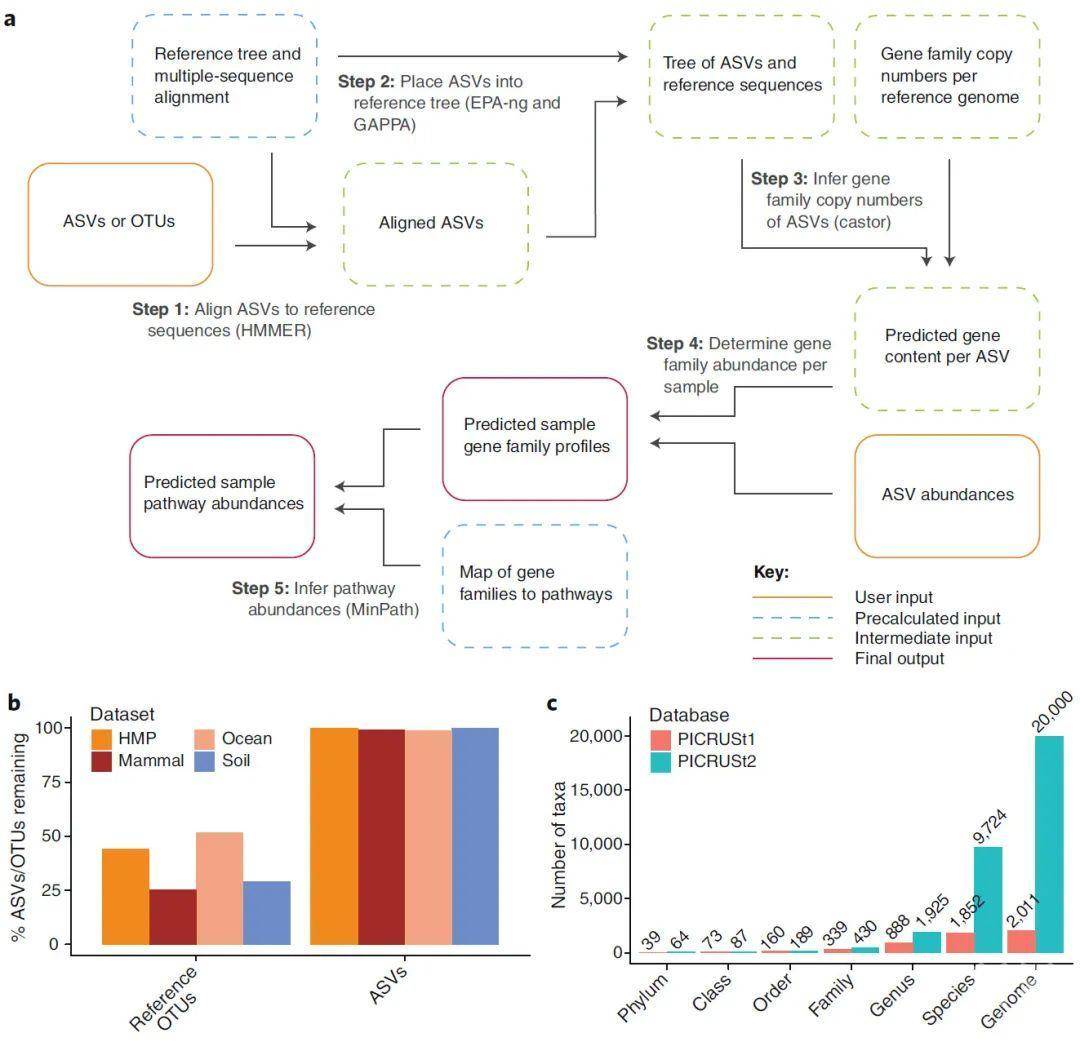
a,PICRUSt2方法由系统发育位置,隐藏状态预测以及按样本的基因和途径丰度列表组成。
将ASV序列和丰度作为输入,并输出基因家族和途径丰度。
PICRUSt2实现中包含默认工作流的所有必需参考树和特征数据库。
b,PICRUSt1流程将预测限制为Greengenes参考数据库中的OTU。
这项要求导致排除了其他16S rRNA基因测序数据集中的许多公开序列。
PICRUSt2放宽了此要求,并且与输入序列是否在参考数据库内无关,这几乎使所有用户的ASV都保留在最终输出中。
c,与PICRUSt1相比,默认PICRUSt2数据库中的物种分类学多样性有所增加,其中种增加近5倍,基因组增加10倍。
PICRUSt2集成了现有的开放源代码工具,以预测环境采样的16S rRNA基因序列的基因组。ASV放置在参考树中,该树用作功能预测的基础。该参考树包含来自整合微生物基因组(IMG)数据库中细菌和古细菌基因组的20,000个完整16S rRNA基因。PICRUSt2中的系统发生放置基于三个工具的输出:HMMER用于放置ASV,EPA-ng确定这些放置的ASV在参考系统发育中的最佳位置,GAPPA用于输出一棵包含ASV放置位置的新树。这样就形成了一个既包含参考基因组又包含环境采样生物的系统树,用于预测每个ASV的个体基因家族拷贝数。对于每个输入数据集,将重新运行此过程,从而允许用户根据需要使用自定义参考数据库,包括可以针对特定微生物生态位的研究进行优化的数据库。
与PICRUSt1中一样,PICRUSt2中使用隐藏状态预测方法来推断采样序列的基因组含量。基本比PICRUSt1中使用的方法更快的R包castor用于核心隐藏状态预测功能。与PICRUSt1中一样,ASV会通过其16S rRNA基因拷贝数进行校正,然后乘以其功能预测值,从而生成预测的基因组。PICRUSt2还提供了每个预测功能的ASV贡献,从而允许进行分类学方面的统计分析。最后,基于结构化的通路映射来推断通路丰度,该映射比PICRUSt1中使用的“基因袋”方法更为保守。
PICRUSt2默认基因组数据库基于IMG数据库7(2017年11月8日)中的41,926个细菌和古细菌基因组,比PICRUSt1使用的2,011个IMG基因组增加了20倍以上。许多其他基因组来自相同物种的菌株,并且具有相同的16S rRNA基因。我们在这些基因组中去除重复的16S rRNA基因,从而产生了20,000个最终的16S rRNA基因簇。PICRUSt2参考数据库的分类学多样性大于PICRUSt1(图1c)。多样性最明显的增加是在物种和属水平上(分别增加了5.3倍和2.2倍),但是所有分类学水平都更加多样化,包括门类,其覆盖范围从39个门增加到64个门(a 1.6 -增加)。
默认情况下支持基于多个基因家族数据库的PICRUSt2预测,包括《京都议定书》中的基因和基因组(KEGG)直系同源物(KO)和酶委员会编号(EC编号)(补充表1)。PICRUSt2通过将最近添加到KEGG数据库中的基因家族包括在内,对PICRUSt1进行了明显的改进。具体而言,与PICRUSt1中的6,909个相比,PICRUSt2中的KO总数为10,543,增长了1.5倍。
我们使用从16S rRNA标记基因和MGS生成的七个已公开数据集中的样本中验证了PICRUSt2的基因组预测。我们使用了三个与人类相关的微生物组数据集:来自喀麦隆个体的57个粪便样本,来自印度个体的91个粪便样本以及来自人类微生物组计划的137个人体的样本。我们还使用了四个非人类相关的数据集,包括77个非人类灵长类动物粪便样本,八个其他哺乳动物粪便样本,六个海洋样本以及22个土壤和蓝莓根际土壤样本。这些数据集显示了序列和环境类型的良好变化(补充表2)。
为每个数据集生成了来自16S rRNA标记基因数据的PICRUSt2 KO预测。我们将这些预测与对应的MGS宏基因组的KO相对丰度进行了比较,这些相对基因丰度是评估预测性能的金标准。我们使用四个替代预测流程进行了相同的分析:PICRUSt1,Piphillin,PanFP和Tax4Fun2。在将所有KO表过滤到所有测试数据库可以输出的6,220个KO之后,计算出与预测KO丰度表和MGS KO丰度表之间匹配的样本的Spearman相关系数(以下称为“相关性”)(图2)。相关度量表示预测数据和观察数据之间KO丰度的等级排序相似性。基于PICRUSt2 KO预测的相关性介于平均值0.79(标准差= 0.028;灵长类动物粪便)至0.88(标准差= 0.019;喀麦隆粪便数据集)之间。对于所有七个数据集,PICRUSt2预测均优于或可与最佳预测方法相媲美(配对样本,双尾Wilcoxon秩和检验P <0.05)。对于非人类相关数据集,基于PICRUSt2预测的相关性要好得多。对于基于参考基因组的环境较差的环境,此结果可能表明基于系统发育的方法相对于基于非系统发育的方法(例如Piphillin)的优势。
图2:PICRUSt2与其他预测软件的结果比较Fig. 2: PICRUSt2 performance characteristics.
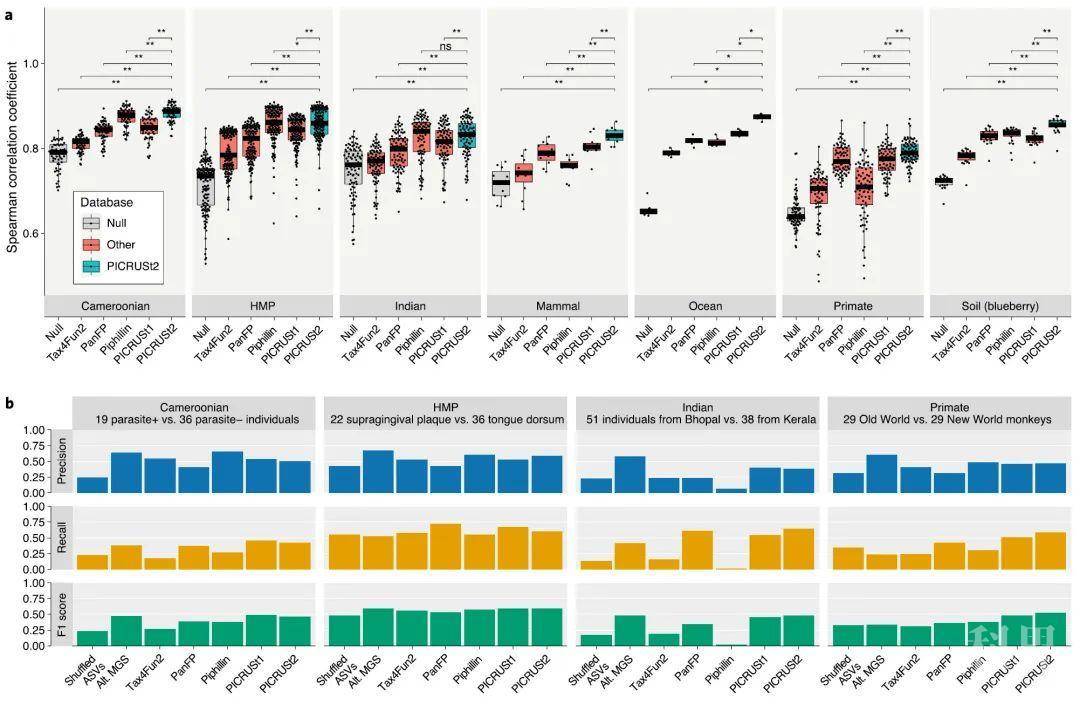
验证PICRUSt2和其他工具KO预测相对于随机宏基因组结果的比较。
a,使用箱线图展示了在不同样本中的预测结果:
喀麦隆人(n = 57),人类微生物组计划(HMP,n = 137),印度人(n = 91),非人类灵长类动物粪便样本(n)的粪便样本(n= 77),其他哺乳动物粪便(n = 8),海水(n = 6)和蓝莓土壤(n = 22)数据集。
配对样本,成对Wilcoxon秩和检验的显著性在每个被测分组的上方显示( P <0.05;* P <0.001;
ns,不显著)。
b,预测的基因组和MGS之间丰度显著差异的KO的比较。
报告每个类别的精确度、召回率和F1得分,并将其与MGS数据进行比较。
精度对应于该类别中重要KO的比例,在MGS数据中也很重要。
召回对应于在该类别中也很重要的MGS数据中重要KO的比例。
F1分数是这些指标的谐波平均值。
所比较的四个数据集的子集显示在每个面板上方。
在通过每个样品拷贝基因的中位数标准化后,对KO相对丰度进行Wilcoxon测试。
显着性定义为错误发现率<0.05。
“按顺序排列的ASV”类别对应于按数据集按顺序排列的ASV标签的PICRUSt2预测。
基因家族经常在基因组中共存,因此样品中缺乏基因家族的独立性可能会限制使用相关性评估基因表相似性(补充图2)。为了解决这种依赖性,我们比较了成对的MGS和预测的宏基因组之间的相关性与MGS功能和有空值(null)参考基因组之间的相关性,该参考基因组由所有参考基因组中的平均基因家族丰度组成。对于所有数据集,PICRUSt2宏基因组表与MGS值相比,与null更相似(图2a)。但是,超出null预期的增加主要是由每个数据集的预测基因组含量(而不是单个样本的含量)驱动的。这些事实实际上仅比在数据集中混用ASV标签时观察到的相关性高出一点(补充图3)。观察到的打乱(Shuffled)的ASV的相关性介于0.77(标准差= 0.196;灵长类动物粪便)至0.84(标准差= 0.178;蓝莓根际)之间。从生物学上讲,这些结果与几种模式是一致的。首先,基因家族在不同分类单元中的拷贝数相关(如Null数据集所捕获)。其次,这些相关性比环境之间的相关性强(如空值和打乱 ASV结果之间的差异所示)。最后,环境之间的差异往往大于环境中样本之间的差异(如PICRUSt2预测与随机ASV结果之间的差异所示)。
验证宏基因组预测的一种补充方法是将16S预测的宏基因组的差异丰度测试结果与MGS数据进行比较。对Piphillin的最新分析表明,在这种方法的基础上,该工具的性能优于PICRUSt2。我们对四个验证数据集的KO预测进行了类似的评估(图2b;参见补充方法和补充结果)。总体而言,与其他预测方法相比,PICRUSt2显示出最高的F1得分,即精确度和召回率的谐波平均值(范围为0.46至0.59;平均值 = 0.51;标准差 = 0.06)。但是,所有预测工具都显示出相对较低的精度,重要的KO的比例在MGS数据中也很重要。特别地,对于PICRUSt2,精确度在0.38至0.58(平均值= 0.48;标准偏差= 0.08)的范围内,对于Piphillin,精确度在0.06至0.66(平均值= 0.45;标准偏差= 0.27)的范围内。在所有情况下,PICRUSt2预测均优于ASV打乱的预测,后者的精度范围为0.22至0.42(平均值= 0.30;标准差= 0.09)。此外,对来自替代MGS处理流程的MGS衍生的KO进行的差异丰度测试仅导致略高的精度(范围从0.57到0.67;平均值 = 0.62;标准差 = 0.04)。综上所述,这些结果突出了利用预测的和实际的宏基因组学数据再现微生物功能性生物标志物的困难。
现在,默认情况下,MetaCyc途径的丰度是PICRUSt2输出的主要高级别的预测。MetaCyc数据库是KEGG的开源替代方案,也是广泛使用的宏基因组学功能分析器HUMAnN2的主要关注点。通过EC基因家族到途径的结构化映射,在PICRUSt2中计算出MetaCyc途径的丰度。与源自MGS的途径相比,这些途径的预测在总体上优于所有指标(配对样本,两尾Wilcoxon秩和检验P <0.05;图3a和补充图4和5)的零分布。与我们之前的分析一样,代表每个数据集内整体功能结构的打乱ASV预测占了该信号的大部分(补充图4)。此外,在这些途径上的差异丰度测试表明,整个数据集和统计方法的F1分数差异很大,而ASV打乱的预测贡献了大部分信号(补充图6)。对于观察到的和ASV打乱的PICRUSt2预测,F1得分分别为0.23至0.62(平均值 = 0.41;标准差 = 0.17)和0.22至0.60(平均值 = 0.34;标准差 = 0.18)。同样,这些结果表明,很难确定鲁棒的差异丰富的全基因组范围的全基因组途径,这突出了一般分析微生物途径的挑战。
图3:PICRUSt2准确预测用于表征整体环境的MetaCyc途径和表型Fig. 3: PICRUSt2 accurately predicts MetaCyc pathways and phenotypes for characterizing overall environments.
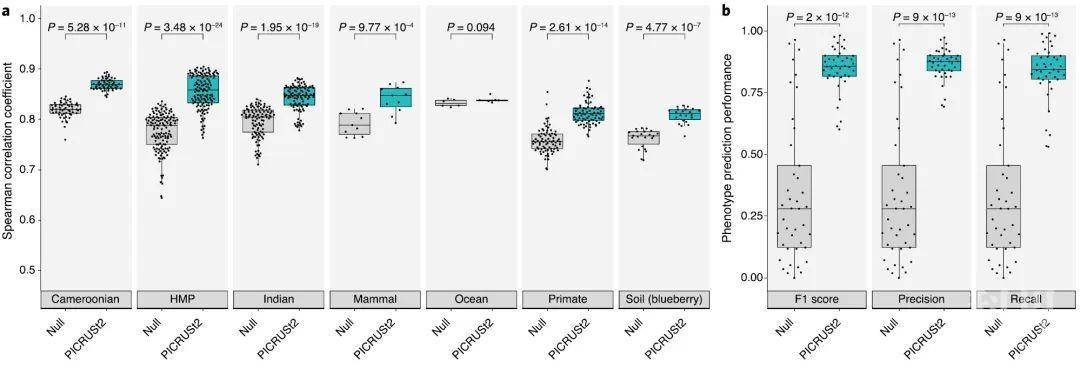
a,PICRUSt2预测的通路丰度与金标准宏基因组测序(MGS)之间的相关性。
显示了每个验证数据集的结果:
喀麦隆人的粪便,人类微生物组计划(HMP),印度个体的粪便,其他哺乳动物粪便,海水,非人类灵长类动物粪便和蓝莓土壤。
这些结果仅限于PICRUSt2和HUMAnN2可识别的575条通路。
b,基于三个指标的二元表型预测性能:
F1得分,准确性和召回率。
每个点对应于测试的41种表型之一。
这里评估的预测是基于分别保留每个基因组,预测该保留基因组的表型以及比较预测值和观察值。
在这种情况下,零分布是基于将参考基因组中的表型随机化并与实际值进行比较,这将为所有三个指标提供相同的输出。
配对样本,成中-Wilcoxon秩和检验的P值显示在每个测试组的上方( P <0.05和* P <0.001)。
在y轴上将截断为低于0.5而不是0,以更好地可视化类别之间的细微差异。
a中的样本大小为57(喀麦隆),137(HMP),91(印度),8(哺乳动物),6(海洋),77(灵长类)和22(土壤)。
现在还可以使用PICRUSt2生成与IMG基因组相关的41种微生物表型的预测。这些代表高水平的微生物代谢活动,例如“葡萄糖利用”和“脱硝剂”,它们被注释为每个参考基因组中存在或不存在。我们进行了一项保留验证,以评估PICRUSt2表型预测的性能,其中涉及将二进制表型预测与每个参考基因组的预期表型进行比较。根据F1分数(平均值= 84.8%; sd = 9.01%),精确度(平均值= 86.5%; sd = 6.21%)和召回率(平均值= 83.5%; sd = 11.4%),这些预测的表现明显优于 零期望值(图3b;Wilcoxon检验P <0.05)。
扩增子功能预测的不足对基于扩增子的功能预测有两个主要的争议。首先是预测偏向于现有参考基因组,这意味着罕见的特定于环境的功能不太可能被识别。随着高质量可用基因组数量的不断增长,该限制随着时间的推移而降低。PICRUSt2还允许将用户指定的基因组用于生成预测,这为研究特定环境提供了灵活的框架。第二个争议是:基于扩增子的预测不能提供分辨菌株特异性功能。这是PICRUSt2和任何基于扩增子的功能预测分析的重要局限性,只能将分类单元区分到它们在扩增的标记基因序列上的差异程度。
PICRUSt2为标记基因的宏基因组推断提供了更高的准确性和灵活性。本文重点讲这些改进,同时还描述了在微生物组研究中确定一致的差异丰富功能的局限性。我们希望PICRUSt2继续扩展功能,让研究者从扩增子测序中得到对功能微生物生态学的见解。
来源:meta-genome 宏基因组
原文链接:https://mp.weixin.qq.com/s?__biz=MzUzMjA4Njc1MA==&mid=2247491081&idx=1&sn=82e71180009b749940c1d0663a3109f0&chksm=fab9f4b8cdce7daec41411595f6d7492569786fbf0f2323afebcc16d76886888fa2584903ea3#rd
版权声明:除非特别注明,本站所载内容来源于互联网、微信公众号等公开渠道,不代表本站观点,仅供参考、交流、公益传播之目的。转载的稿件版权归原作者或机构所有,如有侵权,请联系删除。
电话:(010)86409582
邮箱:kejie@scimall.org.cn



