来源:X一MOL资讯
现代有机合成领域,C-H键官能团化在快速构建分子及其后期修饰中起着越来越重要的作用。然而,多数情况下C-H键的选择性官能团化取决于催化体系对底物上不同电性和位阻的C-H键的区分能力。相比之下,远程C-H键更常见,并且在电性和位阻上几乎无明显的差异,这就给精确控制远程C-H键的官能团化带来了巨大的挑战。
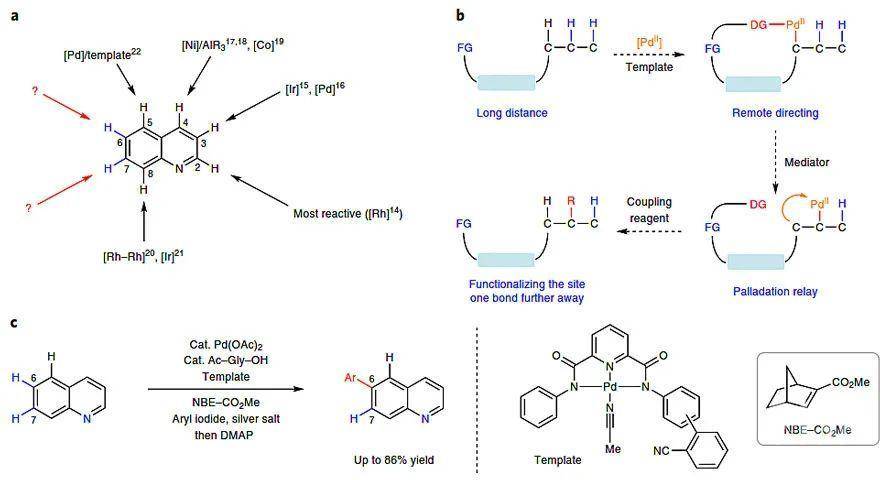
以在药物、农药及材料上有大量应用的喹啉和异喹啉为例,尽管化学家们已经实现了C2-H、C3-H、C4-H、C5-H(远程活化)及C8-H(N协助活化)的活化(下图a),但选择性地进行远程C6-H和C7-H键活化却一直无人成功,这是因为从计算的Fukui指数来看这两个位置的C-H键性质很相似,都是缺电子并且不活泼的。当然,可以通过开发不同几何构造的配体所产生的不同远程导向效应去匹配这两个位置,但这是一个相对“粗放”的想法,意味着要针对不同的底物设计不同的配体,显然实际用途很有限。
这么看来,一个相对靠谱的策略是将远程导向效应和临时的“一键接力”(例如Catellani反应中的降冰片烯)相结合去区分这两个相邻位点的远程C-H键(下图b)。但是,战略再完美也要靠精确有力的战术才能实现。这里还是有几个“关卡”需要一一通过的:首先,弱配位氰基导向的C-H键钯化可能不会与降冰片烯进行接力反应,因为所形成的大环C-H钯化中间体太活泼了,以至于直接同芳基碘代物偶联;其次,降冰片烯参与的接力活化是一个多步骤过程,需要在吡啶或吡啶酮类单齿配体下进行,相反,弱配位的远程活化模板氰基却需要双齿配体;最后,降冰片烯接力后的β-碳消除也依赖于与初次形成的钯-碳键相邻的导向基团,但远程导向基团距离太远以至于无法提供必要的位阻。
最近,美国斯克里普斯研究所(The Scripps Research Institute,TSRI)的余金权教授和加州大学洛杉矶分校(UCLA)的Kendall N. Houk教授等研究者在Nature Chemistry 杂志报道了喹啉/异喹啉类底物远程C6-H和C7-H键的位点选择性官能团化,即利用远程导向模板和降冰片烯对喹啉/异喹啉类底物这两个相邻位点的远程C-H键进行区分和选择性官能团化(下图c)。该反应底物适用性广,官能团耐受性好,有望成为一种通用方法。
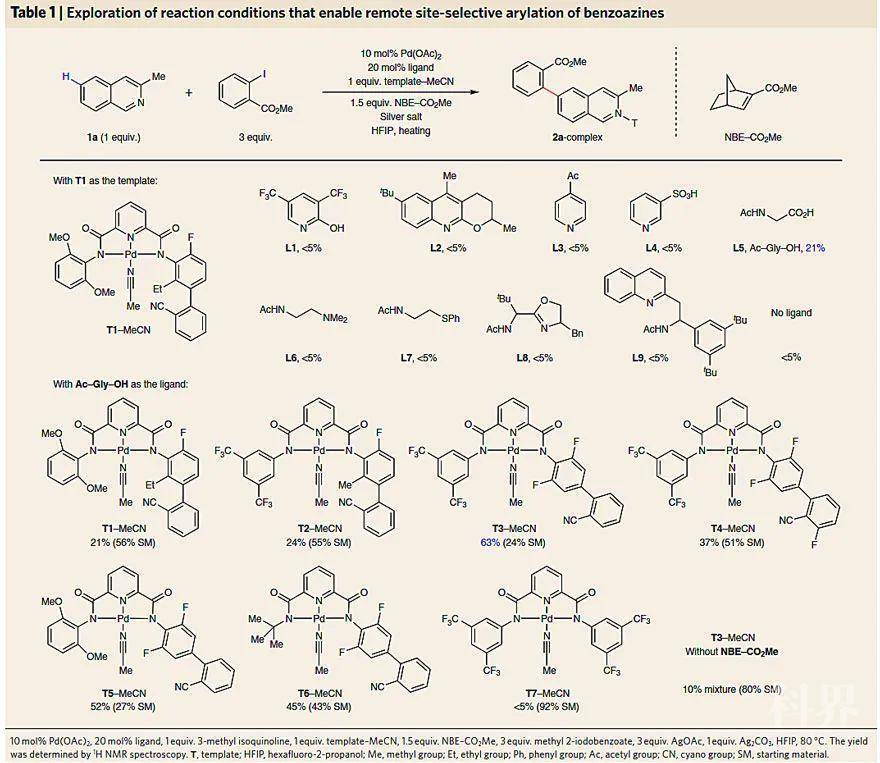
首先,研究人员选择3-甲基-异喹啉为模板底物探索催化体系(下图),结果发现之前在喹啉C5-H键活化中用的导向模板T1与降冰片烯NBE-CO2Me及常与之配合的单齿配体L1-L4并没有“强强联合”的协同效应,这可能是由于吡啶或吡啶酮配体阻止了弱配位的氰基导向基团与二价钯中心结合。
首先,研究人员选择3-甲基-异喹啉为模板底物探索催化体系(下图),结果发现之前在喹啉C5-H键活化中用的导向模板T1与降冰片烯NBE-CO2Me及常与之配合的单齿配体L1-L4并没有“强强联合”的协同效应,这可能是由于吡啶或吡啶酮配体阻止了弱配位的氰基导向基团与二价钯中心结合。
于是,他们尝试使用弱配位的双齿配体(L5-L7)。值得注意的是,当使用L5(Ac–Gly–OH,后称MPAA)时,收率明显的提高(21%)。但使用其他双齿配体L6-L7(弱配位)、L8-L9(强配位)却没有任何改善,这表明需要特殊的配体才能与导向模板和降冰片烯相匹配。接着,他们对模板进行修饰,将T1左边富电子基团换成缺电子基团时(T2),转化率有所提高(24%);而将起导向作用的苯甲腈换到对位时(T3),转化率大幅提高(63%);改变其他结构则导致收率降低(T4-T6)。对照试验表明模板上的氰基以及降冰片烯都是至关重要的。
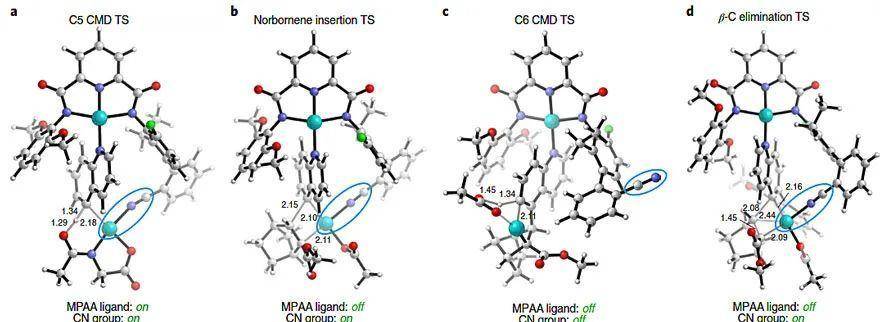
研究人员用密度泛函理论(DFT)进行理论计算,表明这是一种罕见的机理,即导向模版与两个钯金属中心配位。具体来说,在C5位协同金属化-去质子化(concerted metallation–deprotonation,C5 CMD)的过程中,MPAA首先是促进该过程,随后解离,为后续的降冰片烯插入提供空配位点;接着导向的氰基解离进入C6位协同金属化-去质子化;最后通过β-碳消除完成C6位活化。
在上述研究的基础上,研究人员对各种取代的喹啉和异喹啉进行C6位远程活化的底物扩展(下图),结果发现T1适用于C2位无取代的喹啉,T2适用于C2位有取代的喹啉,而T3适用于异喹啉。该反应的底物适用性广,一系列异喹啉衍生物都能够兼容该反应(2a-2d),以中等的收率得到C6-H键官能团化的产物,其中卤素取代的底物还能进行下一步的转化(2b、2d)。对于喹啉类底物,无论底物上带有电中性取代基、供电子取代基还是吸电子取代基(除了带配位能力的氰基2m、阻碍C5-H钯化的甲氧基2n),都能很好的兼容该反应(2f-2q),以中等至较好的收率得到目标产物。此外,双取代的喹啉衍生物以及多环喹啉衍生物都能以中等至良好的收率实现C6-H键官能团化(2r-2v)。值得注意的是,喹喔啉(2w)、苯并恶唑(2x)、苯并噻唑(2y、2z)、吲唑(2aa)和噻吩并吡啶(2ab)也能兼容该反应,同时也能用于喜树碱(可用于抗白血病和抗肿瘤)的后期修饰(2ac)。
研究人员用密度泛函理论(DFT)进行理论计算,表明这是一种罕见的机理,即导向模版与两个钯金属中心配位。具体来说,在C5位协同金属化-去质子化(concerted metallation–deprotonation,C5 CMD)的过程中,MPAA首先是促进该过程,随后解离,为后续的降冰片烯插入提供空配位点;接着导向的氰基解离进入C6位协同金属化-去质子化;最后通过β-碳消除完成C6位活化。
在上述研究的基础上,研究人员对各种取代的喹啉和异喹啉进行C6位远程活化的底物扩展(下图),结果发现T1适用于C2位无取代的喹啉,T2适用于C2位有取代的喹啉,而T3适用于异喹啉。该反应的底物适用性广,一系列异喹啉衍生物都能够兼容该反应(2a-2d),以中等的收率得到C6-H键官能团化的产物,其中卤素取代的底物还能进行下一步的转化(2b、2d)。对于喹啉类底物,无论底物上带有电中性取代基、供电子取代基还是吸电子取代基(除了带配位能力的氰基2m、阻碍C5-H钯化的甲氧基2n),都能很好的兼容该反应(2f-2q),以中等至较好的收率得到目标产物。此外,双取代的喹啉衍生物以及多环喹啉衍生物都能以中等至良好的收率实现C6-H键官能团化(2r-2v)。值得注意的是,喹喔啉(2w)、苯并恶唑(2x)、苯并噻唑(2y、2z)、吲唑(2aa)和噻吩并吡啶(2ab)也能兼容该反应,同时也能用于喜树碱(可用于抗白血病和抗肿瘤)的后期修饰(2ac)。
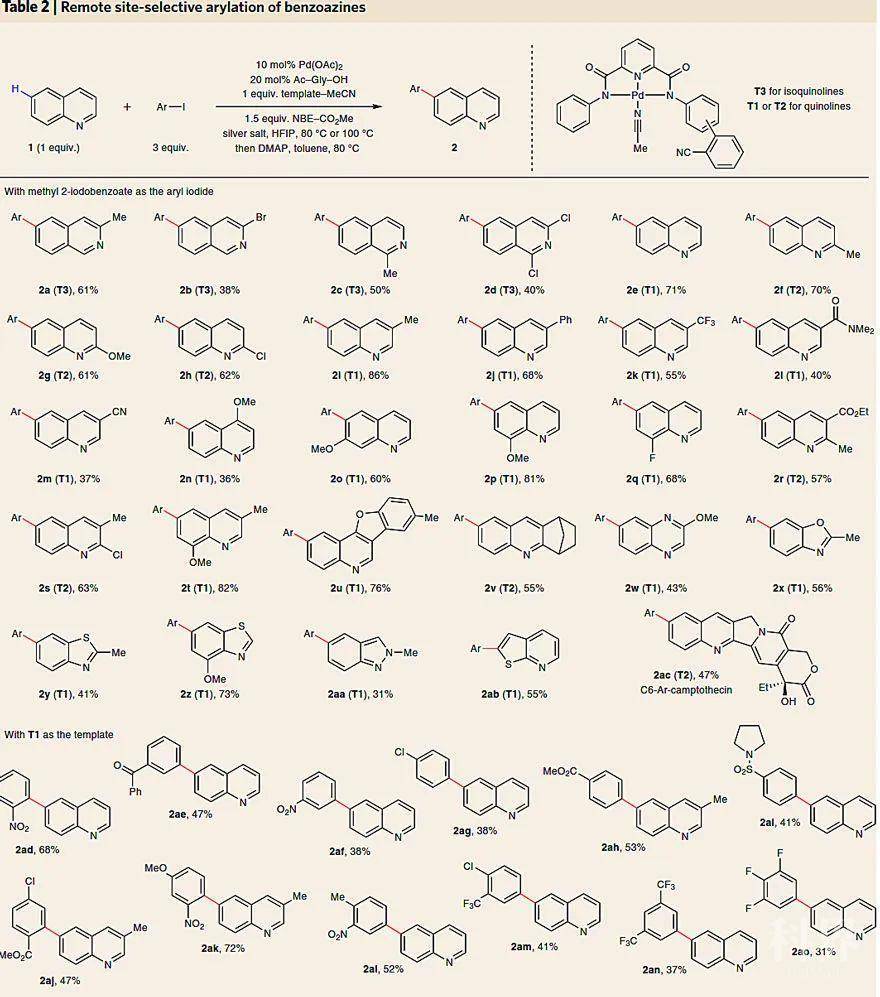
接下来,研究人员对芳基碘化物的底物范围进行了考察。对于单取代的芳基碘化物,无论是邻位、间位还是对位取代(2ad-2ai),都能兼容该反应;二取代或三取代的芳基碘化物都能以中等至较好的收率得到目标产物(2aj-2ao)。然而,富电子的芳基碘化物的收率则较低(< 15%),可能是因为不利于氧化加成。值得一提的是,尽管要使用当量的导向模板,但可以回收再利用。
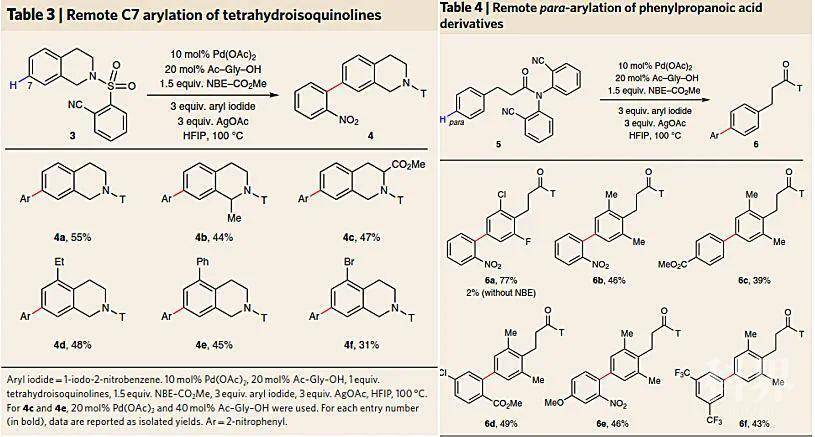
基于上面所讨论的机理,研究人员认为如果底物能模拟导向模板T3的结构,同样也能实现选择性的远程C-H键活化(下图)。他们通过设计含有氰基的U型底物3和5,在不含T3的情况下均能与不同的芳基碘化物进行远程C-H键芳基化。例如:四氢异喹啉类底物能够以中等的收率实现C7位芳基化(4a-4f),且能够耐受各种取代基(如甲基、乙基、酯基、苯基、溴)。此外,该方法也能够实现苯丙酸衍生物的对位芳基化(6a-6f)。
总结
总结
原文:Differentiation and functionalization of remote C–H bonds in adjacent positionsHang Shi, Yi Lu , Jiang Weng , Katherine L. Bay , Xiangyang Chen , Keita Tanaka, Pritha Verma, Kendall N. Houk , Jin-Quan Yu Nat. Chem., 2020, DOI: 10.1038/s41557-020-0424-5
来源:X-molNews X一MOL资讯
原文链接:http://mp.weixin.qq.com/s?__biz=MzAwOTExNzg4Nw==&mid=2657630410&idx=1&sn=ee6da6fe7df84e1c2bb1ed6d7baad186&chksm=80f8111ab78f980c995d2d534ceac27668bf03ead48aa3ee1f94225cb9902435a36838b6e461&scene=27#wechat_redirect
版权声明:除非特别注明,本站所载内容来源于互联网、微信公众号等公开渠道,不代表本站观点,仅供参考、交流、公益传播之目的。转载的稿件版权归原作者或机构所有,如有侵权,请联系删除。
电话:(010)86409582
邮箱:kejie@scimall.org.cn