KRAS 相关信号通路与小分子抑制剂研究进展
[摘要] KRAS 基因属于Ras 基因家族,其编码的 KRAS 蛋白通过激活下游信号通路参与细胞增殖、分化及凋亡等生命活动的调控。某些KRAS 基因突变会引起 KRAS 蛋白过度表达和持续活化,进而过度激活下游信号通路,导致细胞不可控制地增殖恶变,最终造成组织癌变。寻找 KRAS 突变相关肿瘤的治疗策略一直是研究热点之一,然而 KRAS 蛋白突变体是公认的难靶蛋白之一,其直接作用靶点的小分子抑制剂的设计具有很大的挑战性,目前尚无相关药物批准上市。通过对 KRAS 蛋白结构与功能、相关信号通路和突变体在肿瘤发生发展中的作用,以及相关小分子抑制剂的突破性研究进展进行综述,为进一步研发 KRAS 突变肿瘤治疗药物提供参考。
Kirsten 大鼠肉瘤病毒癌基因同源物(kirsten ratsarcoma viral oncogenehomolog,KRAS)蛋白是一个重要的信号传感器,通过与鸟嘌呤三核苷酸磷酸(guanosine triphosphate,GTP)或鸟嘌呤二核苷酸磷酸(guanosine diphosphate,GDP)结合,在活化和失活 2 种状态之间转换,以此来控制细胞信号传导。KRAS 蛋白发生某些突变后,其内在的 GTP 酶(GTPase)活性丧失,因此被锁定在与 GTP 结合的状态即活化状态,从而持续激活下游信号通路,促进肿瘤的发生发展。由于 KRAS 基因突变率高且易诱导肿瘤耐药,因此抑制 KRAS 信号通路被认为可能是肿瘤治疗的有效策略之一。
1 KRAS 简介
大鼠肉瘤病毒同源癌基因(rat sarcoma viraloncogene homolog,Ras)广泛存在于各种真核生物如哺乳动物、果蝇、真菌及线虫中,能够参与调控细胞的增殖、分化、存活及凋亡等生命活动 [1]。人类的 RAS 基因编码 3 种高度同源的 RAS 蛋白:HRAS、NRAS 和 KRAS [2-3]。KRAS 基因位于人类12 号染色体上,长约 35 kb,含有 4 个编码外显子和 1 个 5' 端非编码外显子,其第 4 个外显子有 2 种变异体,可编码 KRAS4A 和 KRAS4B 2 种剪接体 [4-5]。
1.1 KRAS 蛋白结构
KRAS 是一种原癌基因,由其编码 188 ~ 189 个氨基酸组成 KRAS 蛋白,KRAS 蛋白的相对分子质量约为 21 000 [6]。KRAS 蛋白含有 4 个结构域,其中 N 端的第 1 个结构域为 85 个氨基酸残基组成的高度保守序列,第 2 个结构域为相似性相对较低的序列,2 个结构域共同形成对 KRAS 信号传导非常重要的 G 结构域 [7]。G 结构域含有 GTP 结合口袋,该口袋对于下游效应分子与 GTP 酶活化蛋白(GTPase-activating proteins,GAPs)之间的相互作用是必需的。KRAS 蛋白的 C 端含有高变结构区,该结构域在调节蛋白生物活性方面起重要作用 [8]。
1.2 KRAS 蛋白功能
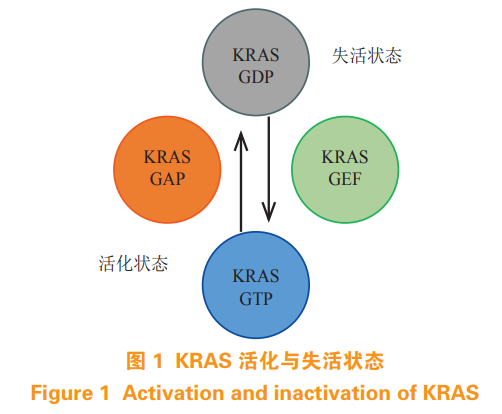
KRAS 蛋白是一类 GTP 结合蛋白,通过与 GTP和 GDP 的结合来调控自身活性(见图 1)。生理条件下,2 种状态的转换受到鸟嘌呤核苷酸交换因子(guanine nucleotide exchange factors,GEFs)和 GTP 酶激活蛋白的调节 [8-9]。GEFs 协助 GTP 取代 GDP 结合并活化 KRAS 蛋白,活化的 KRAS 蛋白进而激活下游信号通路并将信号传递至下游效应器。当 KRAS-GTP 进一步与 GAPs 结合后,KRAS蛋白内在的 GTP 酶活性得到增强,将 GTP 水解成GDP,KRAS 蛋白因而失活 [10-11]。因此,KRAS 蛋白通过与 GTP 或 GDP 结合,在活化和失活状态之间转换,调节下游信号通路的开启和关闭,从而完成信号传导。
2 KRAS 介导的信号通路
KRAS 作为一种信号传感器,可将上游信号分子的刺激信号传递至细胞内,进而调控细胞的增殖、分化、存活和凋亡等生命活动 [12]。KRAS蛋白可被生长因子、趋化因子、Ca2+ 或酪氨酸激酶(tyrosine kinase,TK) 激 活, 活 化 的 KRAS 蛋白可以激活磷脂酰肌醇 3-激酶(phosphoinositide3-kinase,PI3K)-蛋白激酶 B(protein kinase B,AKT)-雷 帕 霉 素 靶 蛋 白(mammalian target ofrapamycin,mTOR)、Ras-快速加速纤维肉瘤(rapidlyaccelerated fibrosarcoma,Raf)-分裂原活化蛋白激酶(mitogen-activated protein kinase,MEK)-细胞外调节蛋白激酶(extracellular regulated protein kinases,ERK)、鸟嘌呤核苷酸解离刺激因子(ral guaninenucleotide dissociation stimulator,RalGDS)- Ras 样蛋白(ras-like protein,Ral)及 Ras 同源蛋白(rashomologue,Rho)/Ras 相关 C3 肉毒素底物(rasrelated C3 botulinum toxin,Rac)等信号通路 [13]。
2.1 PI3K-AKT-mTOR 通路
PI3K-AKT-mTOR 信号通路被认为在细胞增殖、分化、凋亡和葡萄糖转运等细胞生命活动中具有重要作用,同时对肿瘤耐药性的产生也具有较大影响 [14-15]。KRAS 蛋白与膜受体结合激活 PI3K 的p85 亚基,再与 p110 亚基结合使 PI3K 活化,激活的PI3K 催化二磷酸磷脂酰肌醇(phosphatidylinositol4,5-bisphosphate,PIP2)转化成第二信使三磷酸磷脂酰肌醇(phosphatidylinositol 3,4,5-trisphosphate,PIP3)[16]。PIP3 与细胞内含有 PH 结构域的信号蛋白AKT 和磷酸肌醇依赖性蛋白激酶 1(phosphoinositidedependent kinase 1,PDK1)结合,促使 PDK1 磷酸化 AKT 蛋白的苏氨酸磷酸化位点(Thr308),mTOR复合物 2(mTOR complex 2,mTORC2)进一步磷酸化 AKT 蛋白的丝氨酸磷酸化位点(Ser473),从而使AKT 蛋白完全活化 [17-18]。
活化的 AKT 进入细胞核,激活或抑制下游多种效应因子,进而调控细胞的增殖、凋亡及代谢等过程 [19]。一方面,AKT 可直接激活 mTOR 靶蛋白。mTOR 具有丝氨酸/苏氨酸蛋白激酶活性,在调节细胞增殖、存活、代谢及蛋白质合成和转录中起着重要作用 [20]。mTOR 由 mTORC1 和 mTORC2 组成,共同发挥作用 [21]。其中,mTORC1 被 AKT 激活,进而磷酸化真核细胞翻译启动因子 4E 结合蛋白-1(eukaryotic translation initiation factor 4E bindingprotein 1,4EBP-1)和 p70 核糖体蛋白 S6 激酶(p70 ribosomal protein S6 kinase,p70S6K)[22]。磷酸化的翻译抑制因子 4EBP-1 能够促进肿瘤细胞生长相关蛋白的表达,包括细胞周期蛋白 1(cyclin D1)和血管内皮生长因子(vascular endothelial growthfactor,VEGF)[23-24],p70S6K 则可磷酸化并激活核糖体 40S 小亚基 S6 蛋白,进而促进胰岛素受体底物-1(insulin receptor substract-1,IRS-1)的表达,从而导致胰岛素抵抗 [25]。mTORC2 能够阻止蛋白质磷酸酶 2A(protein phosphatae 2A,PP2A)的癌性抑制因子(cancerous inhibitor of protein phosphatase2A,CIP2A)与 PP2A 的结合,促使 PP2A 对原癌基因 c-Myc 的去磷酸化而使其降解,从而降低促凋亡 miRNA 的转录及其对 E2F 转录因子 1(E2Ftranscription factor 1,E2F1)的表达抑制,从而抑制细胞凋亡 [26]。另一方面,AKT 还可以磷酸化并激活 Bcl- xl /Bcl-2 相关死亡启动子(Bcl-xL/Bcl-2-associated death promoter,BAD),活化的 BAD 进而与伴侣蛋白 14-3-3 结合, 使 BAD 蛋白去磷酸化以及与 Bcl-2/Bcl-xl 的结合受到抑制,从而抑制细胞凋亡 [27-28]。
2.2 Ras-Raf-MEK-ERK 信号通路
Ras-Raf-MEK-ERK 信号通路是一条有丝分裂原活化蛋白激酶(mitogen-activated protein kinase,MAPK)通路,能将细胞外信号传递入细胞核内,从而参与调控细胞的增殖、分化、迁移、免疫应答及转录等生命活动 [29-30]。
KRAS 蛋白结合并活化其膜受体,使之与生长因子受体结合蛋白 2(growth factor receptor-boundprotein 2,Grb2) 和 SOS (son of sevenless)蛋白结合,形成受体-Grb2-SOS 三元复合物。该复合物的 SOS 与其受体或受体底物的酪氨酸磷酸化位点结合后,再与 KRAS-GDP 结合,促使 GTP 取代 GDP而激活 KRAS [31],形成的 KRAS-GTP 将胞浆中的Raf 蛋白募集到质膜上,并诱导 Raf 构象改变,促进 Raf 同源或异源二聚化而激活 [32]。Raf 的 C 端催化域与双特异性激酶 —— MEK1/2 结合,并使之磷酸化而活化。MEK1/2 进而磷酸化并激活 ERK1/2,活化的 ERK 可以磷酸化 p90 核糖体 S6 激酶(ribosomalS6 kinase,RSK)、血清反应因子(serum response factor,SRF)、E26 转录因子(E26 transformationspecific transcription factors,ETS)及 Elk-1 转录因子(ETS like-1 protein)等来调节相应靶基因的转录翻译,从而参与调控细胞增殖、分化及迁移等生命活动 [33-35]。
2.3 RalGDS-Ral 通路
RalGDS 是一种 GTP/GDP 交换因子,能促进Ral 蛋白的 GDP/GTP 转 换,RalGDS 是 Ras 的 下游信号蛋白,能够通过与 Ras 的活性形式结合而发挥作用 [36]。首先,活性形式的 Ras-GTP 通过与RalGDS 的 Ras/Rap 结 合 结 构 域(Ras/Rap bindingdomain,RBD)结合将其递送至质膜上与 Ral 结合,从而使 Ral 活化 [37]。Ral 蛋白的下游效应因子包括Rac/ 细胞分裂周期蛋白 42(cell division cycle 42,Cdc42)、TANK 结合激酶 1(TANK binding kinase1,TBK1)和动力蛋白相关蛋白 1(dynamin-relatedprotein 1,Drp1)等 [38]。其中,Rac/Cdc42 属于 Rho家族 GTP 酶,其被认为与细胞的迁移运动有关,因此 KRAS 突变蛋白通过下游信号通路激活 Rac/Cdc42 能够促进肿瘤细胞的迁移 [38]。TBK1 不仅能够介导Ⅰ型干扰素的产生和抗病毒先天免疫,也能够介导 KRAS 依赖性肿瘤细胞的生存和增殖 [39]。Drp1 不仅是线粒体和过氧化物酶体裂变所必需的,而且能够促进 KRAS 驱动的糖酵解通量,从而促进肿瘤生长 [40-41]。
2.4 Rho/Rac 通路
Rho 小 G 蛋白家族属于 Ras 超家族,是一类具有 GTP 酶活性的核苷酸依赖型的 GTP 结合蛋白,包括有 Rho、Rac、Cdc42、Rnd、RhoD 及 TTF 等亚家族成员,Rho 小 G 蛋白通过与 GDP 和 GTP 结合在活化和失活状态之间转换,这 2 种状态的转换由 GEFs、GAPs 和鸟苷酸解离抑制剂(guaninenucleotide dissociation inhibitors,GDIs)3 种蛋白共同调节,从而调控细胞增殖、骨架形成及细胞周期等生命活动 [42-43]。GEFs 和 GAPs 对 Rho 小 G 蛋白的调节作用与 KRAS 蛋白一样,而 GDIs 能够通过抑制核苷酸的解离来干扰 Rho 家族蛋白与 GDP和 GTP 结合 [44]。活化的 Rho 蛋白能够激活 Rho 相关激酶(Rho-associated kinase,ROCK)和威奥综合征蛋白(Wiscott-Aldrich syndrome protein,WASP)。其中,ROCK 具有丝氨酸 / 苏氨酸蛋白激酶活性,能够介导细胞死亡 [45]。WASP 通过与WASP 相互作用蛋白(WASP-interacting-protein,WIP)相互作用调节 T 细胞抗原受体(T cell antigenreceptor,TCR)的信号传导,从而抑制 T 细胞淋巴瘤生长 [46]。
3 KRAS 基因突变与肿瘤发生发展
3.1 KRAS 基因突变
Ras 基因是人类癌症中首个被鉴定出来的致癌基因 [46],该家族基因突变约占所有肿瘤相关突变的20%~30%,与人类恶性肿瘤的发生发展密切相关[47]。KRAS 基因突变约占 Ras 基因家族突变的 85%,并比 Ras 家族其他基因突变更易促进肿瘤的发生 [48]。遗传学和生物化学研究表明,在 Ras 突变中,KRAS突变频率显著高于 HRAS 和 NRAS 突变 [49]。点突变是最常见的 KRAS 基因突变方式,一般发生在 2 号外显子第 12、13 位甘氨酸密码子及 3 号外显子第61 位谷氨酰胺密码子上,尤以第 12 位密码子突变最为常见 [50],已发现的 12 位甘氨酸(G)点突变包括 G12A、G12D、G12V 及 G12C 等,其中 G12D突变频率远高于其他 G12 突变 [51]。
3.2 KRAS 基因突变与肿瘤发生发展
KRAS 基因发生突变后,其编码表达的 KRAS蛋白的构象发生改变,使 KRAS 蛋白几乎完全丧失内在的 GTPase 酶活性,GTP 从 KRAS 上解离减少,GDP 与 KRAS 结合能力减弱,从而使 KRAS 蛋白一直处于活化状态,持续激活下游信号通路,导致细胞内的信号转导出现紊乱,造成细胞不可控制地增殖恶变,进而促进肿瘤的发生 [52]。
3.2.1 胰腺癌 胰腺癌是一种常见的消化系统恶性肿瘤 [53],KRAS 突变是胰腺癌患者中最常见的突变类型,超过 90% 的胰腺癌患者存在 KRAS 突变,说明KRAS 基因突变对胰腺癌的发生发展具有重要的影响 [54-55]。
Cheng 等 [56] 研究发现,KRASG12D 突变与胰腺癌组织中调节性 T 细胞(regulatory cells,Tregs)高比例浸润密切相关,KRASG12D 突变通过激活下游的 MEK/ERK 通路,促进 ERK 磷酸化,造成白细胞介素 10(interleukin-10,IL-10)和转化生长因子-β(TGF-β)的过表达,使得 Tregs 大量富集,最终导致胰腺癌组织产生免疫抑制。Wang 等 [57] 通过KRAS 基因敲除实验发现,利用 shRNA 敲除 KRAS导致人胰腺癌细胞中的白血病抑制因子(leukemiainhibitory factor,LIF)mRNA 的 表 达 下 降, 且 发现抑制 KRAS 下游蛋白 MEK 能够下调人胰腺癌细胞中 LIF 的表达,而过表达的 LIF 能够以反馈机制介导 KRAS 驱动肿瘤的癌变。Liou 等 [58] 发现KRASG12D 突变可诱导产生线粒体氧化应激,引发胰腺腺泡细胞的去分化,并激活蛋白激酶 D1(proteinkinase D1,PKD1)/ 核因子 κB(nuclear factor κB,NF-κB)通路而上调表皮生长因子受体(epidermalgrowth factor receptor,EGFR)的表达,导致驱动胰腺癌癌前病变的形成,并且 PKD1 作为 PI3K/Akt 通路的重要信号蛋白,其异常活化会影响 Akt 的完全活化,从而影响 Akt 下游蛋白的表达。此外,Liang等 [59] 发现腺苷二磷酸核糖基化因子 6(adenosinediphosphate ribosylation factor 6,ARF6) 是 KRAS的下游信号蛋白,KRAS 突变能通过 MEK 信号通路传导激活 ARF6。ARF6 除能够以反馈机制维持KRAS 介导的 ERK 激活外,还能够增强胰腺癌侵袭组织和逃避免疫系统的能力 [60]。可见,KRAS 突变能够通过信号通路传导激活多种下游信号蛋白来促进胰腺癌细胞的增殖、侵袭转移及耐药等。
3.2.2 结直肠癌 结直肠癌(colorectal cancer,CRC)是一种常见的消化道恶性肿瘤,其中转移性结直肠癌患者的 5 年生存率不足 12%,约有 40% 的转移性结直肠癌患者伴有 Ras 基因突变,而其中最主要突变是 KRAS 突变 [61-63]。
KRAS 基因突变后,激活下游 Ras-Raf-MEKERK 信号通路,进而破坏树突状细胞(dendriticcells,DC)的募集功能和主要组织相容性复合体Ⅱ(major histocompatibility complex Ⅱ,MHC Ⅱ )的分子呈递能力,从而诱导结肠细胞癌变 [64-65]。Brandt 等 [66] 研究揭示,KRASG12V 突变能够细胞特异性地活化 ERK 激酶,且 ERK 活化同时受到细胞特异性的 MEK/ERK 信号通路调节,导致 ERK 激酶高度活化。此外,KRAS 突变蛋白可协同 β-连环蛋白利用高度活化的 ERK 激酶促进小肠隐窝细胞扩增。Pierroa 等 [67] 研究发现,KRASG13D 突变通过重构 ERK 通路依赖的基质相互作用分子(stromalinteraction molecule,STIM)的表达来影响 ERK 通路介导下游信号分子的表达,从而抑制钙池操纵的 Ca2+ 流 入(store-operated Ca2+ entry,SOCE) 和钙释放激活钙离子(Ca2+ release-activated current,CRAC)通道,使细胞内 Ca2+ 释放减少,最终促进结直肠癌细胞的增殖和迁移。Chu 等 [68] 研究发现,KRAS 突变体(KRASG13D 和 KRASG12V)能够通过激活 MEK-ERKs 介导的信号通路上调 Y-box 结合蛋白 1(Y-box binding protein 1,YB-1)的表达,进而激活胰岛素样生长因子 -1 受体(insulin-like growthfactor-I receptor,IGF-IR) 蛋白的表达,从而促进结直肠癌的肝转移。Toda 等 [69] 研究发现,KRASG13D突变激活肿瘤细胞中 PI3K-Akt-mTOR 信号通路,诱导天冬酰胺合成酶(ASNS)的过表达,导致天冬氨酸水平显著降低和天冬酰胺的含量水平上升,使肿瘤细胞适应在缺乏营养条件下生长。隋华等 [70] 研究发现,NF-κB 是 PI3K/Akt 信号通路的下游蛋白,KRAS 突变后,PI3K/Akt 通路被激活,磷酸化增加,使得进入细胞核内的 NF-κB 增多,进而上调 P-糖蛋白(P-glycoprotein,P-gp)的表达,导致结直肠癌细胞耐药。相关研究表明,KRAS 突变能够通过介导 PI3K、MEK 及 ERK 等蛋白的信号通路参与结直肠癌细胞的增殖、转移及耐药等问题。
3.2.3 非小细胞肺癌 肺癌是一种常见的恶性肿瘤,可分为非小细胞肺癌(non-small cell lung cancer,NSCLC) 和 小 细 胞 肺 癌(small cell lung cancer,SCLC), 其 中 NSCLC 占肺癌的 80% ~ 85%[71]。尽管近年对肺癌免疫治疗及靶向药物的研究取得一定进展,但晚期 NSCLC 患者 5 年生存率仍不足15%。多种基因突变可诱发 NSCLC,其中 KRAS 基因突变占 20% ~ 30%[72-73]。KRAS 突变可通过多条信号通路和多种效应因子诱导肿瘤细胞的增殖、恶变和耐药等促进肺癌的进展,尤其是对 NSCLC 的发生发展具有重要影响。
KRAS 突变主要通过促进肿瘤细胞增殖,减少肿瘤细胞凋亡,促进肿瘤血管生成、浸润和转移及促进肿瘤细胞耐药等方面来促进 NSCLC 的发生发展。Sparmann 等 [74] 研究发现,ERK-MAPK 通路的激活能够上调 IL-8 的表达,KRAS 突变能诱导 IL-8的过表达,从而驱动 NSCLC 细胞的增殖恶变。Wu等 [75] 和 Park 等 [76] 研究发现,KRAS 能够通过激活 ERK1/2、c-Jun 氨基末端激酶(c-Jun N-terminalkinase,JNK)及 PI3K 等信号蛋白介导的信号通路来诱导尿激酶型纤溶酶原激活物(urokinasetype plasminogen,uPA) 及 其 受 体(urokinase-typeplasminogen activator receptor,uPAR)的表达,从而降解细胞外基质(extracellular matrix,ECM),导致肿瘤细胞不可控的增殖、浸润及转移。Shi 等 [77] 利用野生型 KRAS 过表达和 KRAS12D 突变的人和小鼠细胞,发现突变 KRAS 显著上调 miR-30c 和 miR-21,并通过抑制 NF1、RASA1、BID 和 RASSF8 等关键的肿瘤抑制基因诱导产生耐药性、促进细胞侵袭和迁移。Tao 等 [78] 通过构建 KRASG12D 肺癌小鼠模型发现,KRASG12D 的过表达激活 ERK 信号通路,并以正调控机制激活核转录因子 2(nuclear factor 2,Nrf2)通路,从而增加细胞保护性应激反应基因Nrf2 的转录,促使肿瘤细胞产生耐药性。Riquelme等 [79] 研究发现,与其他 KRAS 突变相比,KRASG12C突变能够激活 PI3K/AKT 信号通路,而激活的 AKT能够磷酸化肿瘤细胞中的 zeste 基因增强子同源物 2(enhancer of zeste homolog 2,EZH2),从而促进肿瘤细胞的侵袭转移。
4 KRAS 突变蛋白的小分子抑制剂
当与 GTP 结合时,活化状态的 KRAS 蛋白表面的 Switch-I 口袋(S-IP)和 Switch-II 口袋(S-IIP)处于“关闭”状态,即无法与小分子抑制剂结合,且其与 GTP 具有强亲和力,细胞中高浓度的 GTP导致靶向 GTP 结合位点的竞争性抑制剂难发挥作用。由于 KRAS 蛋白结构表面平滑,除 GTP 结合位点外,难以寻找能够与小分子抑制剂产生有效结合的位点 [80-82]。研究人员曾试图开发与 KRAS 效应器结合或靶向 KRAS 核苷酸结合位点的小分子抑制剂,以破坏 KRAS 的 Switch-I 和 Switch-II 域活性构象、促进 GEF 的作用 [83-85],但所得化合物在亲和力、生物活性和成药性方面均不尽人意。随着 KRAS 突变体结构研究的深入,近年来研究人员通过巧妙途径设计获得直接靶向 KRAS 突变体的小分子抑制剂。
4.1 ARS-853 和 ARS-1620
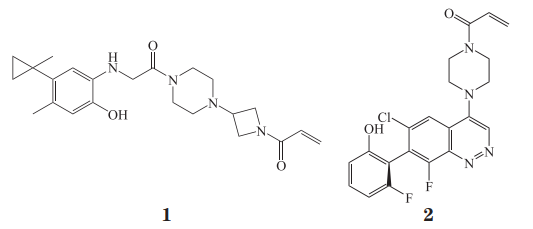
Ostrem 等 [86] 开创性地提出靶向 KRAS 蛋 白的 S-IIP 进行小分子抑制剂设计。基于该思路,Wellspring Biosciences 公司研究人员筛选到靶向 GDPKRASG12C 复合体的小分子抑制剂 ARS-853(1),该化合物通过共价作用结合于复合体中KRAS的S-IIP,使其更倾向于与 GDP 结合,将其锁定在“失活”构象,显著抑制 H358 细胞增殖(IC50=1.6 μmol·L-1)[87-88]。
Janes 等 [89] 对化合物 1 进行结构优化,得到细胞活性是其 10 倍的喹唑啉化合物 ARS-1620(2),其 IC50 为 150 nmol· L-1。共晶研究结果显示,与化合物 1 相比,化合物 2 增加额外关键作用,其 1位 N 原子与 KRASG12C 组氨基酸残基 His 95 形成氢键,使 4 位弹头与 KRASG12C Cys 12 以不可逆结合方式形成的刚性构型更稳定。化合物 2 特异性抑制人NSCLC 细胞 NCl-H358、人胰腺癌细胞 MIA-PaCa2和人大细胞肺癌细胞 LU65 内的 KRASG12C 蛋白,并显著抑制这 3 种肿瘤细胞的增殖。该化合物具有良好的口服生物利用度(F > 60%)和血浆稳定性,在 MIA-PaCa2 细胞和 H358 细胞皮下异种移植小鼠胰腺癌和肺癌模型实验中,以剂量依赖性方式抑制肿瘤生长,随着给药剂量从 200 mg · kg-1 增至400 mg·kg-1,KRASG12C 突变肿瘤的体积显著缩小 [89]。
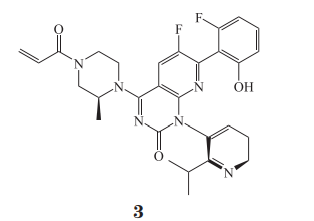
4.2 AMG-510
与 Wellspring Biosciences 公司不同,安进公司瞄准 KRAS 蛋白表面隐藏的凹槽进行抑制剂设计。化合物 AMG-510(3)与 KRASG12C 突变蛋白Cys 12 发生不可逆结合,将 KRAS 蛋白锁定在与 GDP结合的非激活状态,从而特异性地抑制携带 G12C突变的 KRAS 蛋白 [90-91]。与化合物 2 相比,化合物3 的芳香环与 KRAS 上隐藏在 His 95 位置的表面凹槽结合,增强与 KRASG12C 蛋白的相互作用力,其对NCI-H358 细胞的 KRASG12C 的结合速率即表观速率常数 kobs ·[I]-1 为(1.1×104±1.4×103)mol·L-1·s-1,从而使得细胞活性显著提高(NCI-H358 细胞,IC50 =6 nmol·L-1;MIA PaCa-2 细胞,IC50=9 nmol·L-1)[92]。
化合物 3 能够抑制 KRASG12C 信号传导, 选择性地降低 KRASG12C 突变细胞系的活力,而对其他 KRAS 突变细胞系无影响 [93]。在 KRASG12C 肿瘤细胞小鼠模型中,化合物 3 能够增强炎性分子趋化因子的表达,从而显著抑制肿瘤生长 [92]。Ⅰ/Ⅱ期临床试验结果显示,化合物 3 对局部晚期/晚期KRASG12C 突变 NSCLC、结直肠癌和 SCLC 的疗效良好,患者总疾病控制率达到 90%[94]。
4.3 MRTX-849
MRTX-849(4) 由 Mirati Therapeutics 公司设计,结合模式与化合物 3 相似,通过共价方式与KRASG12C 的 S-IIP 口袋中的 Cys 12 不可逆地结合,并将其锁定在非活性 GDP 结合状态,将 KRASG12C蛋白不可逆地锁定在“关闭”状态,从而阻断KRAS 信号传导 [95-96]。
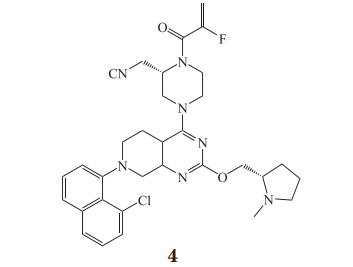
化合物 4 能抑制 KRASG12C 突变癌细胞系中KRAS 下游 ERK 和核糖体蛋白 S6 的磷酸化,对绝大多数 KRASG12C 突变细胞系都具有明显的生长增殖抑制作用(2D 培养,IC50 为 10 ~ 973 nmol· L-1;3D 培养,IC50 为 0.2~1 042 nmol·L-1),且对 KRASG12C突变蛋白的选择性是野生型 KRAS 蛋白的 1 000 倍以上。体内实验结果显示,化合物 4 可剂量依赖性减小多种肿瘤细胞的异种移植瘤小鼠的肿瘤生长,包括人肺癌细胞 H2030 和 H2122,人 NSCLC 细胞SW1573 和 H358,人食管鳞癌细胞 KYSE-410,人胰腺导管腺癌细胞 MIA PaCa-2[97]。
5 结语与展望
鉴于 KRAS 突变体的结构特点、其介导信号通路的复杂性以及 KRAS 突变型肿瘤的耐药性,KRAS 一度被称为“不可成药靶点”。近年来,随着 KRAS 突变体结构研究的深入,相关药物研发已取得重大进展,研究人员巧妙地设计出一些具有广阔前景的 KRAS 突变体小分子抑制剂,但目前仍无小分子药物被批准上市。KRAS 突变型肿瘤治疗依然面临诸多问题,例如 KRAS 突变型肿瘤容易耐药。有研究者提出开发 KRAS 上游或下游信号通路抑制剂、协同致死基因抑制剂、肿瘤疫苗及免疫治疗等新策略,并希望能通过联合用药的方式解决 KRAS突变型肿瘤耐药问题。
来源:药学进展
版权声明:除非特别注明,本站所载内容来源于互联网、微信公众号等公开渠道,不代表本站观点,仅供参考、交流、公益传播之目的。转载的稿件版权归原作者或机构所有,如有侵权,请联系删除。
电话:(010)86409582
邮箱:kejie@scimall.org.cn