肌萎缩性嵴髓侧索硬化症(ALS)是一种致命的神经退行性疾病,患者表现为运动神经元丧失。ALS 的病理机制尚不清楚,血脑屏障受损已被认为是疾病发病和进展的一个可能因素。
ALS 和 NMOSD 是两种罕见的神经系统疾病,近年来的研究表明,ALS 和 NMOSD 的患病率分别为3.43/10万和2.56/10万。因此,ALS 和 NMOSD 共存的情况极为罕见,目前只有2例报道,1例NMOSD 发生在 ALS 之前,另一例长期存在的 ALS 后来发生了贯穿性脊髓炎。
最近,韩国淳春大学医院报告一位女性 ALS 患者,在 ALS 诊断时携带AQP4抗体,但没有表现出NMOSD症状,但ALS被诊断出6年以后被诊断出NMOSD 。以下为该病例的详细情况:
患者在53岁时首次出现左上肢无力。肌肉无力缓慢进展到双侧上肢和下肢,深部腱反射增加,但没有疼痛或感觉症状。实验室影像学研究和电诊断进行肌萎缩侧索硬化症的诊断。大脑和脊柱磁共振成像(MRI)没有发现异常。除 AQP4抗体外,实验室检查均正常。
55岁起被诊断为肌萎缩性侧索硬化症。由于病情恶化,她卧床不起,接受了气管切开术。她上肢很虚弱,指屈肌和伸肌的力量达到医学研究委员会(MRC)2级,腿部肌肉达到 MRC 3级。
59岁时,神经系统检查显示除双手指呈 MRC 二级外,其余四肢均有明显的软弱和萎缩,MRC 等级为0。T4区有感觉水平,下肢有感觉丧失。足底伸肌反应出现在两侧。同时伴随膀胱功能障碍。脊髓的磁共振成像显示整个脊髓的T2超强度。
脑脊液分析显示轻度淋巴细胞增多,蛋白水平升高,脑脊液白细胞计数为7/uL,脑脊液蛋白为267.06 mg/dL。自身免疫性血管炎、副肿瘤抗体、血清抗体和病毒感染的脑脊液 PCR 检测均为阴性,但 AQP4抗体为阳性。
根据 NMOSD 的国际共识诊断标准,她被诊断为血清阳性 NMOSD。在静脉注射1000毫克甲基强的松龙5天后,疼痛有所缓解,但腿部无力没有改善。出院一个月后,门诊随访检查未见好转,双腿瘫痪。进一步的结果无法评估。
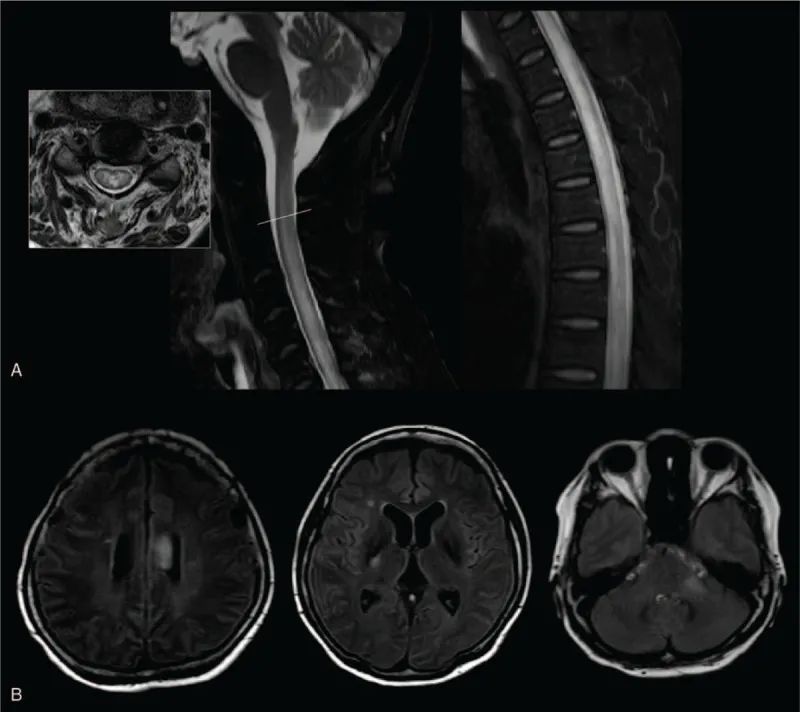
肌萎缩侧索硬化症是一种进行性运动神经元疾病,在疾病的晚期,由于其本身的严重缺陷,其他疾病可能无法被发现。这个病人已经有严重的虚弱,但是疼痛,感觉丧失和膀胱功能障碍可能是诊断贯穿性脊髓炎的线索,这是 NMOSD 的开始。
因此,此病例的报道提示:在临床诊断中,如果 ALS 患者有 AQP4抗体,则建议仔细跟进 NMOSD 的风险。
参考资料:
Kim JY, Oh HJ, Kim Y, Seok JM. Sporadic amyotrophic lateral sclerosis with seropositive neuromyelitis optica spectrum disorder: A case report. Medicine (Baltimore). 2021;100(16):e25580.
来源:梅斯
原文链接:http://mp.weixin.qq.com/s?__biz=MzI0Njc5ODM4MQ==&mid=2247553212&idx=3&sn=f96506ec7512f777d08a2f353cd17e32
版权声明:除非特别注明,本站所载内容来源于互联网、微信公众号等公开渠道,不代表本站观点,仅供参考、交流、公益传播之目的。转载的稿件版权归原作者或机构所有,如有侵权,请联系删除。
电话:(010)86409582
邮箱:kejie@scimall.org.cn



