
▲第一作者:刘加利
通讯作者:卫智虹,焦海军
通讯单位:中国科学院山西煤炭化学研究所,山西大学,莱布尼茨催化研究所
论文DOI:10.1021/acscatal.0c05562
全文速览
本工作在基于脂肪族PNP钳形钴配合物催化羰基化合物加氢反应的理论研究中取得重要进展。基于密度泛函理论研究了脂肪族PNP钳形钴(I/III)配合物之间的相互转换反应,并系统研究了其催化羰基化合物(CH2O、PhCHO、CH3COCH3、PhCOCH3、PhCOOCH3)加氢反应过程中的稳定性、真正的活性物种以及优势反应机理。理论研究结果表明催化剂相互转换反应的最低能量路径存在自旋交叉,且羰基化合物加氢反应的机理和底物性质以及反应条件相关。能量性质相关分析表明,醛酮底物的H–转移能垒ΔG≠(H–)和底物的氢负离子亲和势以及醛酮酯底物的H+转移的逆反应能垒ΔG≠,–(H+)和产物的脱质子能存在较好的线性关系。
背景介绍
发展廉价3d金属催化体系是实现绿色化学和可持续发展的重要途径,因而受到广泛关注。然而,3d金属配合物催化剂存在稳定性差、反应活性低等问题。多齿配体可极大提高催化剂的稳定性,且金属–配体双功能作用也提高了廉价金属配合物催化剂的催化活性,配体的辅助作用也使得H–转移和H+转移分别存在outer–sphere/inner-sphere和innocent/non-innocent机理的竞争。利用脂肪族PNP钳形配体([HN(CH2CH2PR2)2]),已实现Fe、Mn、Co高活性催化体系的开发。其中,脂肪族PNP钳形钴配合物催化剂由于具有多样的氧化态、自旋态和配位化学环境,其电子转移可以通过单电子和双电子方式进行(Figure 1),且多数钴配合物具有顺磁性,为配合物的表征和反应机理的研究带来极大困难。而理论计算能克服实验困难,从电子层面系统地研究不同氧化态、自旋态、几何构型的脂肪族PNP钳形钴催化剂的稳定性和相互转换及反应条件下催化活性物种与优势反应机理。
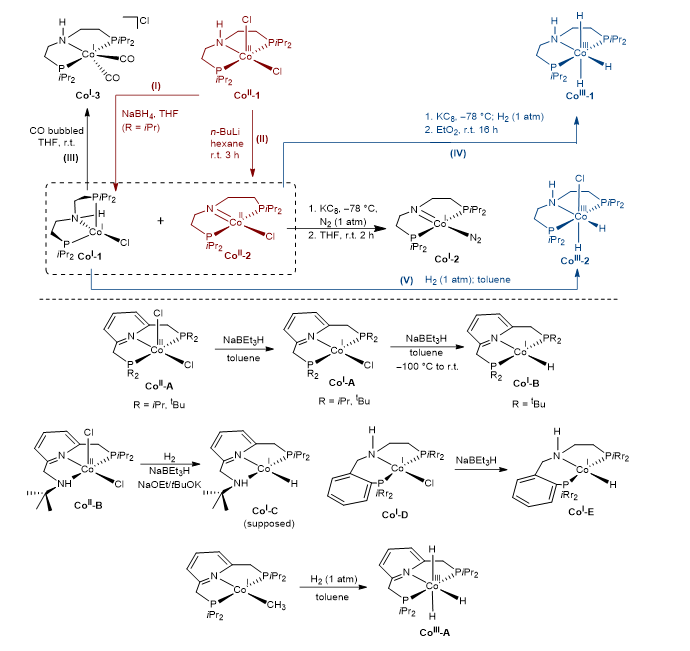
▲Figure 1. Reported Co-pincer complexes in exchangeable oxidation states
本文亮点
(1)相同价态的钴基amine(2CoI/2CoIII)和amido(1CoI/1CoIII)配合物的相互转化过程存在自旋交叉(Figure 2)。
(2)羰基化合物加氢反应的H–转移考虑了传统的inner-sphere和双功能outer-sphere机理,H+转移考虑了配体N–H只提供氢键稳定作用的innocent和N–H发生可逆质子转移的non-innocent机理。
(3)羰基化合物加氢反应的H–转移和H+转移逆反应吉布斯自由能垒分别与底物的氢负离子亲和势和产物脱质子自由能存在较好的线性关系。
(4)揭示了醛酮酯底物加氢反应过程中真正的活性物种,并发现羰基化合物加氢反应的机理取决于反应物的性质与反应条件。
图文解析
(1)Figure 2的结果表明CoIII配合物的基态是三重态,CoI配合物的基态是单重态。单重态11CoIII和三重态31CoI的转化反应以及单重态12CoIII和三重态32CoI的转化反应都存在自旋交叉。
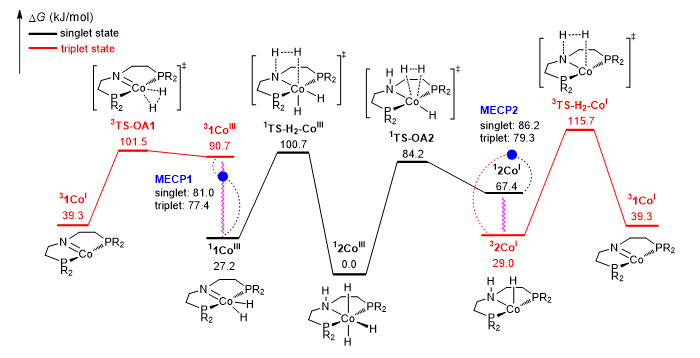
▲Figure 2. Minimum energy path of the CoIII and CoI complexes interconversion (R = isopropyl)
(2)由于催化剂相互转换反应的最高能量(115.7 kJ/mol)(Figure 2)和已报道的加氢反应的能垒相近。我们以羰基化合物为底物,分别计算了基于CoIII和CoI催化循环的加氢反应能垒和优势机理。结果表明两种催化循环下H+转移的优势路径都是氢键稳定的innocent机理;基于CoIII催化循环的H–转移遵循outer-sphere机理;除CH+COCH3底物外,基于CoI催化循环的H–转移遵循更有利的inner-sphere机理。且2CoI催化剂对于羰基化合物的加氢催化活性高于2CoIII。
(3)基于独立的CoIII和CoI催化循环,分别研究了H–和H+转移吉布斯自由能垒与底物的氢负离子亲和势和醇盐的质子亲和势、醇脱质子能的关系。结果表明,H–转移能垒ΔG≠(H–)和底物的氢负离子亲和势ΔGH- (S)的线性相关性较低(Figure 3a,b)。这是由于PhCOOCH3存在较大的偏离,进一步能量分解分析表明PhCOOCH3加氢过渡态结构中催化剂和底物的变形能较大。对于H+转移反应,醇盐R1R2CHO-获得质子生成醇的质子亲和势PA和ΔG≠(H+)相关性较低(Figure 3 c,d),而H+转移反应的逆反应能垒∆G≠,-(H+)和醇脱质子自由能ΔGDH+ (S)存在非常好的线性关系(Figure 3e,f)。因此,文章预测可以利用底物的氢负离子亲和势和产物脱质子自由能作为描述符预测H–/H+转移能垒。
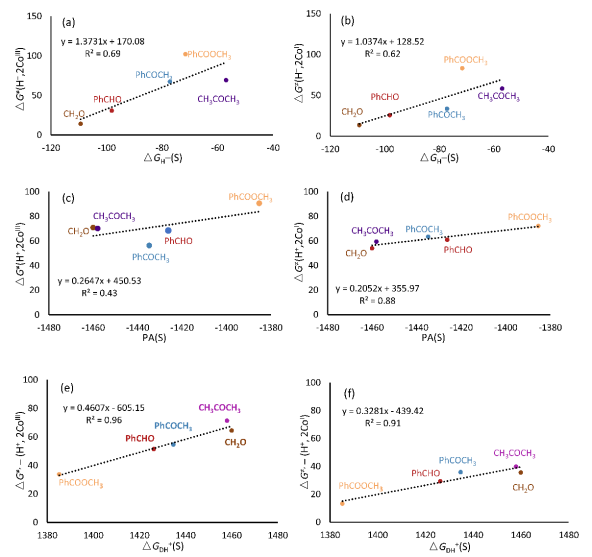
▲Figure 3. Correlation between ∆G≠ for hydride transfer and hydride affinity of substrate catalyzed by 2CoIII (a) and 2CoI (b); ∆G≠ for proton transfer and PA value of R1R2CHO- form of product alcohols catalyzed by 2CoIII (c) and 2CoI (d) as well as ∆G≠ of alcohol deprotonation and deprotonation ∆G of alcohol (R1R2CHOH) catalyzed by 2CoIII (e) and 2CoI (f).
(4)基于理论计算,综合考虑CoIII和CoI催化循环,如Figure 4所示,对于底物CH2O,PhCHO和PhCOCH3,加氢反应能垒低于催化剂相互转换的能垒,因此CoI和CoIII催化循环可以独立进行,两种价态的催化剂都是可能的活性物种。PhCOOCH3加氢反应能垒比催化剂相互转换的能垒高,真正的催化剂是CoIII物种。对于CH3COCH3加氢反应,CoI和CoIII催化循环相互竞争,催化循环取决于反应条件。以上的研究表明羰基化合物催化加氢反应机理取决于底物与反应条件。
▲Figure 4. Simplified most favorable Gibbs energy profile for CH2O, PhCHO, CH3COCH3, PhCOCH3 and PhCOOCH3 hydrogenation based on CoI/CoIII catalytic cycle (* for value labeled by obtained via outer-sphere mechanism)
总结与展望
本文利用密度泛函理论系统研究了脂肪族PNP钳形钴配合物催化剂在羰基化合物加氢反应中的稳定性,阐明了CH2O、PhCHO、CH3COCH3、PhCOCH3、PhCOOCH3加氢反应中真正的催化活性物种及反应机理,为实验条件优化提供理论依据。此外,加氢反应能垒和反应物、底物能量性质的线性相关性分析为构建反应能垒的描述符奠定了基础。
作者介绍
卫智虹,山西大学分子科学研究所,讲师、硕士生导师。2019年博士毕业于德国Rostock大学莱布尼茨催化研究所(LIKAT),师从焦海军研究员。同年加入山西大学李思殿教授团队,主要研究方向为主族化学、金属有机化学及均/多相催化,目前在Angew. Chem. Int. Ed.、Nat. Commun, ACS Catal.、 ACS Cent. Sci.、J. Catal.、Chem. Sci.、J. Phys. Chem. C、Catal. Sci. Technol.、Chem. Eur. J.、等国际期刊发表SCI论文30余篇。
来源:研之成理
原文链接:http://mp.weixin.qq.com/s?__biz=MzUxMDMzODg2Ng==&mid=2247563270&idx=3&sn=8b8c5cfe8561a60bebf50e15466be966
版权声明:除非特别注明,本站所载内容来源于互联网、微信公众号等公开渠道,不代表本站观点,仅供参考、交流、公益传播之目的。转载的稿件版权归原作者或机构所有,如有侵权,请联系删除。
电话:(010)86409582
邮箱:kejie@scimall.org.cn




