来源:X一MOL资讯
最近西班牙瓦伦西亚理工大学化工技术研究所(ITQ)Avelino Corma课题组在ACS Catalysis 上连续发表两篇论文[1-2]。有趣的是,这两篇研究论文都是实验和理论的结合,而且研究的对象都是和分子筛材料有关系。但是,两篇论文涉及的反应和催化材料又有显著的差异。因此,笔者将简要的介绍两篇论文,看看顶尖的催化专家的研究思路。
第一篇文章是关于小孔分子筛催化甲醇制烯烃(MTO)反应。对于MTO反应,大连化物所刘中民院士团队的DMTO技术已经取得了商业化上的巨大成功。但是从这个反应本身来说,其复杂的机理并没有被彻底的研究清楚。因此,相关的基础研究工作依然在进行中,并有多个知名的中外课题组活跃其中。最近加州理工大学的Mark Davis组对一系列的小孔分子筛进行了研究,并且提出不同分子筛的孔结构可以和MTO反应的产物分布进行关联(ACS Catal., 2019, 9, 6012)。但是,这篇论文并没有完全阐述清楚分子筛孔结构是如何影响烯烃产物的分布情况。一般认为,MTO反应都是通过“烃池”(hydrocarbon pool)机理来发生;而烃池的形成有和分子筛的孔结构和酸性位点的分布有直接的关系。对于烃池里面的中间态物种,不同的催化剂可能也会有差异,从而导致对不同的烯烃选择性。这篇论文的思路就是,通过研究中间体分子和分子筛骨架的相互作用来试图从分子筛结构影响中间体结构这个思路去解释催化行为(见图1)。
图1. 通过考察MTO反应的中间体和分子筛骨架的相互作用来解释分子筛结构对MTO反应选择性的影响。图片来源:ACS Catal.
具体来说,作者首先计算了甲基化的六元环中间体在不同结构的小孔分子筛(CHA、AEI、RTH、ITE)中和甲醇的反应过程,并分析各种可能的路径以及不同中间体的稳定性。结合过去的文献以及作者自己的计算结果,他们把MTO反应的关键的中间物种锁定在五甲基苯基正离子(g5MBa+)和七甲基苯基正离子(g7MB+)上。同时,通过一系列的计算和分析,作者引入了一个参数来作为衡量标准,也就是g5MBa+和g7MB+两个中间体的能量差值(Eint(7/5)),因为这个差值直接决定了MTO反应中,paring路径和side-chain路径的比例,也就会直接影响产物中C3-C4产物与C2产物的比例。这两个中间体的稳定性也和分子筛的骨架结构有关。
为了验证上述计算结果,作者合成了一系列的小孔分子筛,并且测试了MTO的反应性能。通过将理论计算结果和MTO反应性能进行关联,作者发现,Eint(7/5)越大,那产物中丙烯含量就越高。并且两种烯烃(C3=/C2=)的比例和Eint(7/5)有比较好的线性关系(图2)。
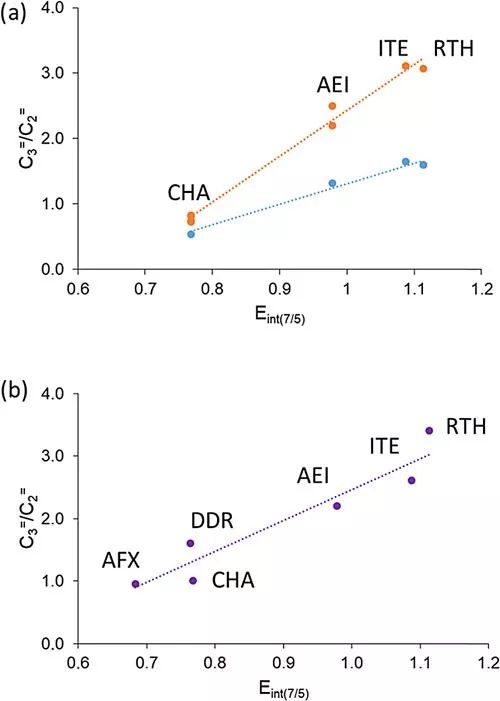
图2. 理论计算结果中提炼出来的参数(Eint(7/5))和实验中丙烯/乙烯比例进行关联。Eint(7/5)数值越大,丙烯的选择性越高,反应越倾向于paring路径。图片来源:ACS Catal.
第二篇文章是关于分子筛限域的Pt团簇在低温条件下催化NO+CO反应(140-200 K)。和上一篇文章相反,这一篇文章从实验结果出发,然后用理论计算结果来解释实验现象。首先,作者合成了三个负载型Pt催化剂,分别是原子级分散的Pt-SA样品,含有亚纳米Pt团簇的Pt-CL样品以及包含Pt纳米粒子的Pt-NP样品(如图3所示)。作者搭建了一套原位红外和质谱联用的装置(operando IR)来实时的跟踪固体催化剂的表面吸附物种以及反应产物。通过这套装置,作者研究在极低温度下,不同尺寸的Pt物种在催化NO+CO反应过程中的演化行为,从而探讨在这个反应中金属物种的尺寸效应。(需要强调的是,这篇文章的出发点是基础研究,所以在如此的低温下,转化率都非常低)
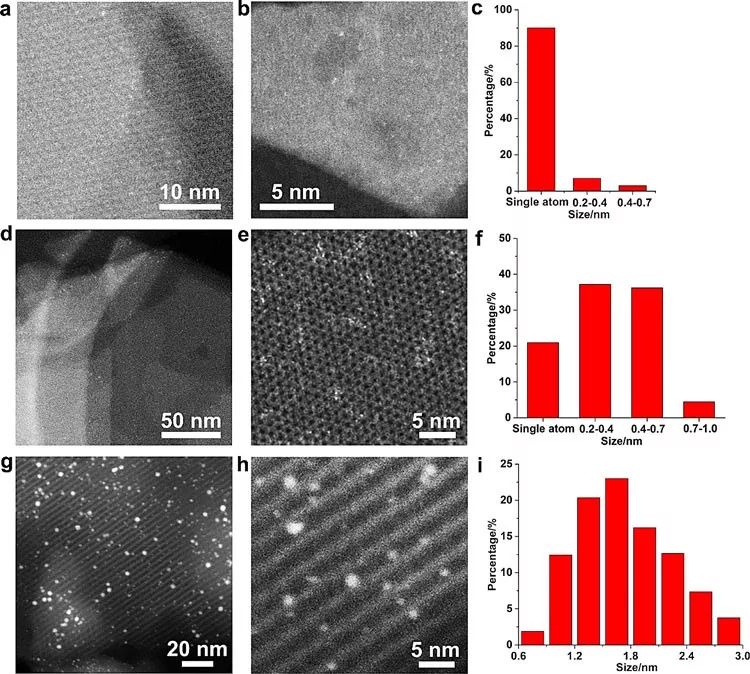
图3. 不同尺寸的Pt催化剂,从单原子到团簇到纳米粒子。图片来源:ACS Catal.
如图4所示,作者发现,对于Pt-SA样品,在NO+CO反应中,该催化剂在初始状态并没有表现出活性,而是经过一个几百秒的诱导期,从而产生CO2和N2。然后作者发现,在反应后的样品中,产生了Pt的团簇。这说明,在反应条件下,原子级分散的Pt物种会发生团簇。而这种团簇行为也被DFT计算证实了。作者发现,在相同的条件下,如果用含有Pt团簇或者Pt纳米粒子的催化剂进行上述反应,就能在初始阶段产生CO2和N2。这些结果表明,团聚态的Pt应该是该反应的活性物种。同时,作者还利用原位IR光谱对吸附在Pt物种表面的各个物种进行分析,并且进行了一些动力学方面的研究。并且,作者还发现,反应温度也会成为一个影响不同尺寸Pt物种催化行为的重要因素。
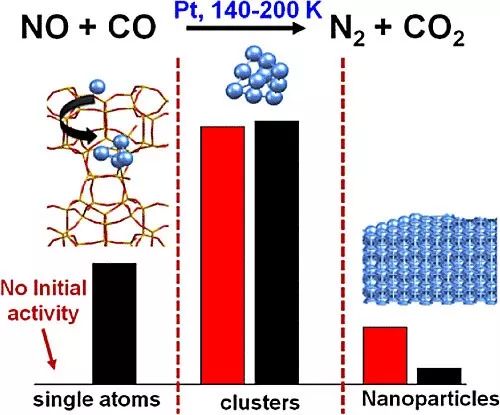
图4. 三种不同类型的负载型Pt催化剂在NO+CO反应中的性能。图片来源:ACS Catal.
为了解释观察到的实验现象,作者进行了DFT计算。首先,从NO+CO这个模型反应的基元步骤出发,作者分析了这个反应的一些关键步骤和可能几种反应路径,最后确定了一条最有可能的路径,包括NO分子的解离成N和O原子,然后是CO吸附在Pt位点上,与O反应,变成CO2。最后,N原子结合成N2。作者计算了在不同尺寸的Pt物种上的各个步骤的能垒,并分析了不同尺寸的Pt物种上的决速步骤(如图5所示)。
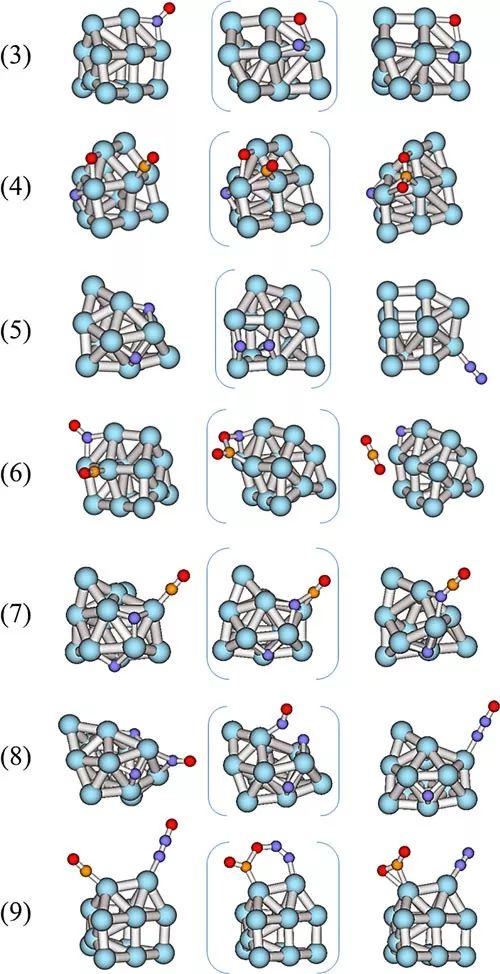
图5. 通过DFT计算来研究Pt团簇催化NO+CO反应的机理。图片来源:ACS Catal.
简单来说,实验和理论结果吻合的较好。对于低温下的NO+CO反应,NO的解离需要多个Pt原子来协同实现。但是,不同尺寸的Pt物种的氧化还原能力不同,这就导致解离后的N和O吸附在Pt表面,与CO的反应性能不同。Pt纳米粒子倾向于被NO氧化,然后很难被CO还原。同时,对于N2的生成,也是一个关键步骤。当反应温度从140 K升高到200 K,Pt纳米粒子又对CO有较强的吸附能力,使得表面被CO“毒化”。所以综合来看,Pt的团簇本身独特的氧化还原性能导致其在这个反应中较好的活性。
从上述两个工作中可以看到,无论是面对典型的酸催化反应(MTO)还是氧化还原反应(NO+CO),如果要从基元反应步骤这个层次去理解反应过程都是需要付出巨大的努力。这也不是一两篇文章能彻底解决的问题。但是,Corma教授课题组的工作表明,实验和理论可以比较好的结合起来,针对实验中遇到的一些新奇的现象或者说难以通过实验手段去验证的想法,用理论计算来进行研究;或者是理论计算中发现了一些“可能的、有趣的”现象,可以设计相关的实验区进行验证。这种计算驱动的实验设计在这两个工作中都有展示。总之,实验和理论可以相辅相成、互相佐证。需要特别强调的是,在这两个工作中,理论计算都尝试去从分子-原子层面理解反应的机理,而不仅仅是简单的用理论计算结果去“解释”催化剂的活性顺序。
来源:X-molNews X一MOL资讯
原文链接:http://mp.weixin.qq.com/s?__biz=MzAwOTExNzg4Nw==&mid=2657623735&idx=2&sn=966310fca2d780dae8b7cc3cbcd00808&chksm=80f83767b78fbe71c26d03cb662e6c782e6b2bb9911e7a6708e62786bf50e3bf809b8db70485&scene=27#wechat_redirect
版权声明:除非特别注明,本站所载内容来源于互联网、微信公众号等公开渠道,不代表本站观点,仅供参考、交流、公益传播之目的。转载的稿件版权归原作者或机构所有,如有侵权,请联系删除。
电话:(010)86409582
邮箱:kejie@scimall.org.cn




