来源:BioArt植物
N6-甲基腺嘌呤 (N6-methyladenosine, m6A) 是一种广泛存在的RNA修饰,在调控拼接和翻译等方面具有重要的作用 (Wang et al., 2015, Xiao et al., 2016)。目前基于抗体检测的方法(m6A-seq)得到广泛使用 (Luo et al., 2014),但该方法无法精确到单碱基的水平。m6A-CLIP (Ke et al., 2015) 和miCLIP (Linder et al., 2015) 可以从单碱基精度检测m6A位点,然而由于文库构建流程繁琐,导致应用受限。不依赖于抗体的方法m6A-REF-seq (Zhang et al., 2019) 和MAZTER-seq (Garcia-Campos et al., 2019) 能从单碱基精度识别和定量ACA基序上的RNA修饰,极大的促进了RNA修饰的研究。但如果想检测ACA基序之外的其它已知的DRACH(D=G/A/U,R=G/A,H=A/U/C)修饰还需要开发更为全面和便捷的单碱基水平定量方法。目前牛津纳米孔技术 (ONT) 的直接RNA测序 (DRS) 技术具有检测RNA中碱基修饰信号的潜力,但目前还缺乏对应的基于DRS直接在单碱基和单转录本水平定量检测m6A修饰的方法。
2021年1月7日,福建农林大学海峡联合研究院林学中心顾连峰教授课题组在Genome Biology期刊在线发表了题为Quantitative profiling of N6-methyladenosine at single-base resolution in stem-differentiating xylem of Populus trichocarpa using Nanopore direct RNA sequencing的研究论文。该研究提供了一种可在单转录本单碱基水平的分辨率定量m6A修饰的方法,为在动植物中的m6A修饰研究提供了一种极为有效的检测手段。利用该方法首次在杨树次生木质部中定量描述选择性多聚腺苷酸化和m6A修饰的关联。该研究首先检测人工合成的有m6A修饰和无m6A修饰位点周围的Nanopore直接RNA测序输出的原始电流信号,提取其信号均值、中值、标准差及宽度等特征构建XGBoost模型,测试数据集的结果表明其m6A修饰预测精度达97%。
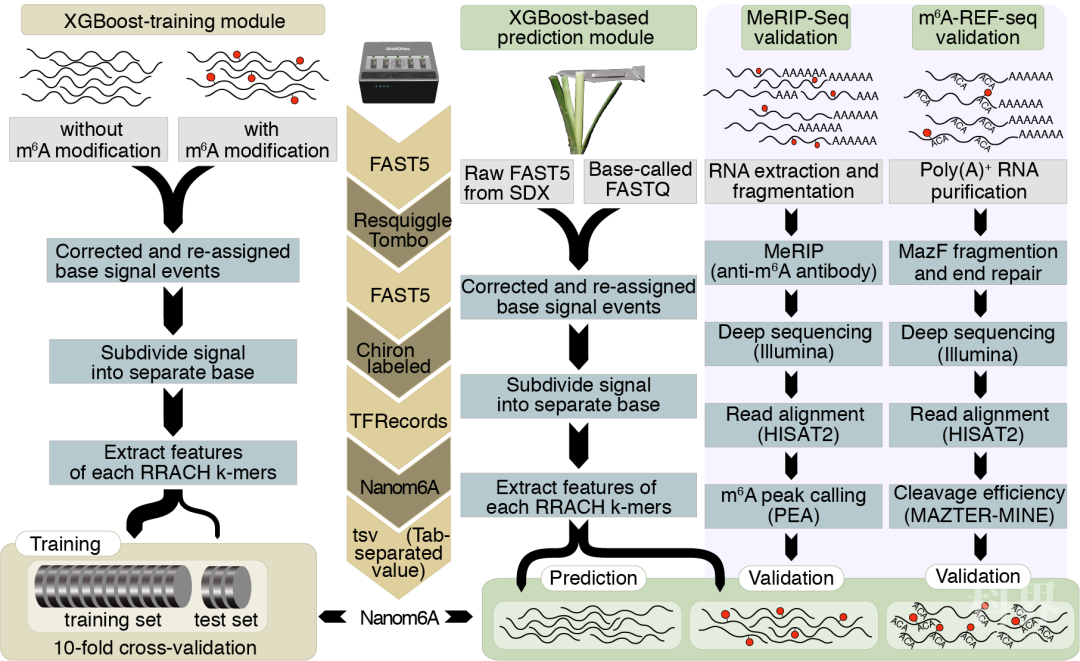
为了验证这种方法的准确性,首先使用该方法对人类HEK293T细胞系和shMETTL3的DRS数据中进行m6A位点识别,并和已发表的基于ligation-assisted extraction and thin-layer chromatography (SCARLET) (Liu et al., 2013) 技术获得marker基因进行比较。发现可以准确检测到TUG1、TPT1、ACTB及BSG这些RNA上的已知m6A修饰位点,这些位点大多位于DRS读段的3’末端区域。基于DRS获得的野生型和METTL3突变体上的m6A修饰率,与MeRIP-Seq的结果完全吻合,并且突变体的修饰率整体上均呈现降低趋势,说明该m6A定量识别方法可以应用在动物的DRS数据上。
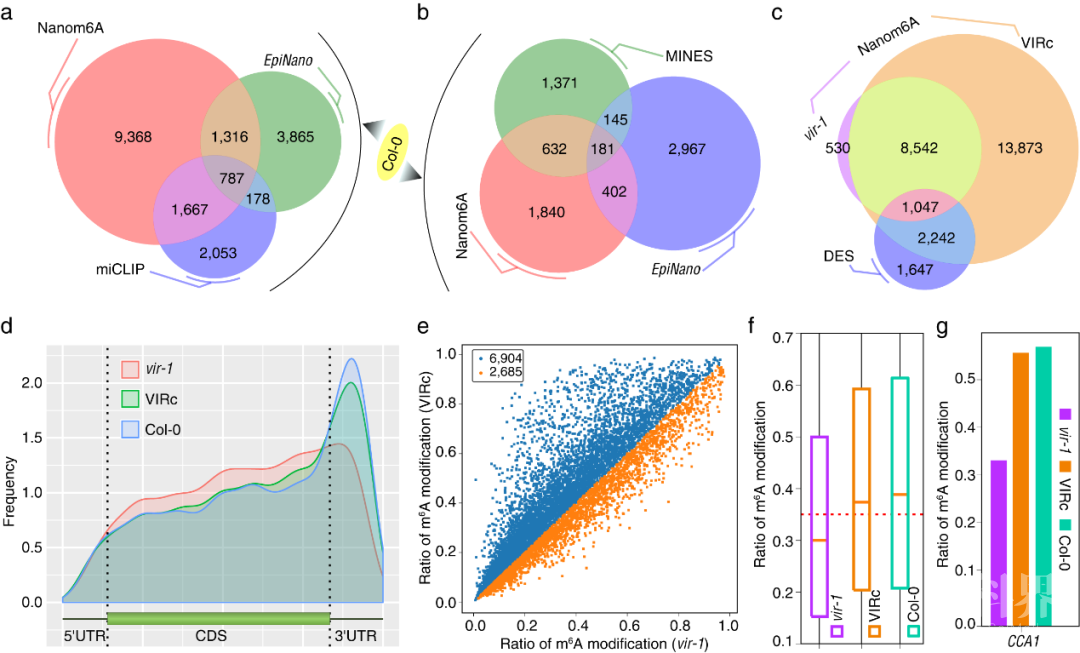
为了进一步验证该方法的通用性,对Parker等报道的拟南芥野生型、VIRILIZER(vir-1)突变体和VIRc回补株系的DRS数据(Parker et al., 2020)采用本文方法进行m6A修饰识别,显示在vir-1突变体中m6A位点明显减少,从整体分布上看在vir-1突变体中m6A位点在终止密码子和3’UTR区域明显减少。因此通过植物DRS数据获得修饰信息和遗传学证据一致,说明该m6A定量识别方法也可以应用在植物的DRS数据上。
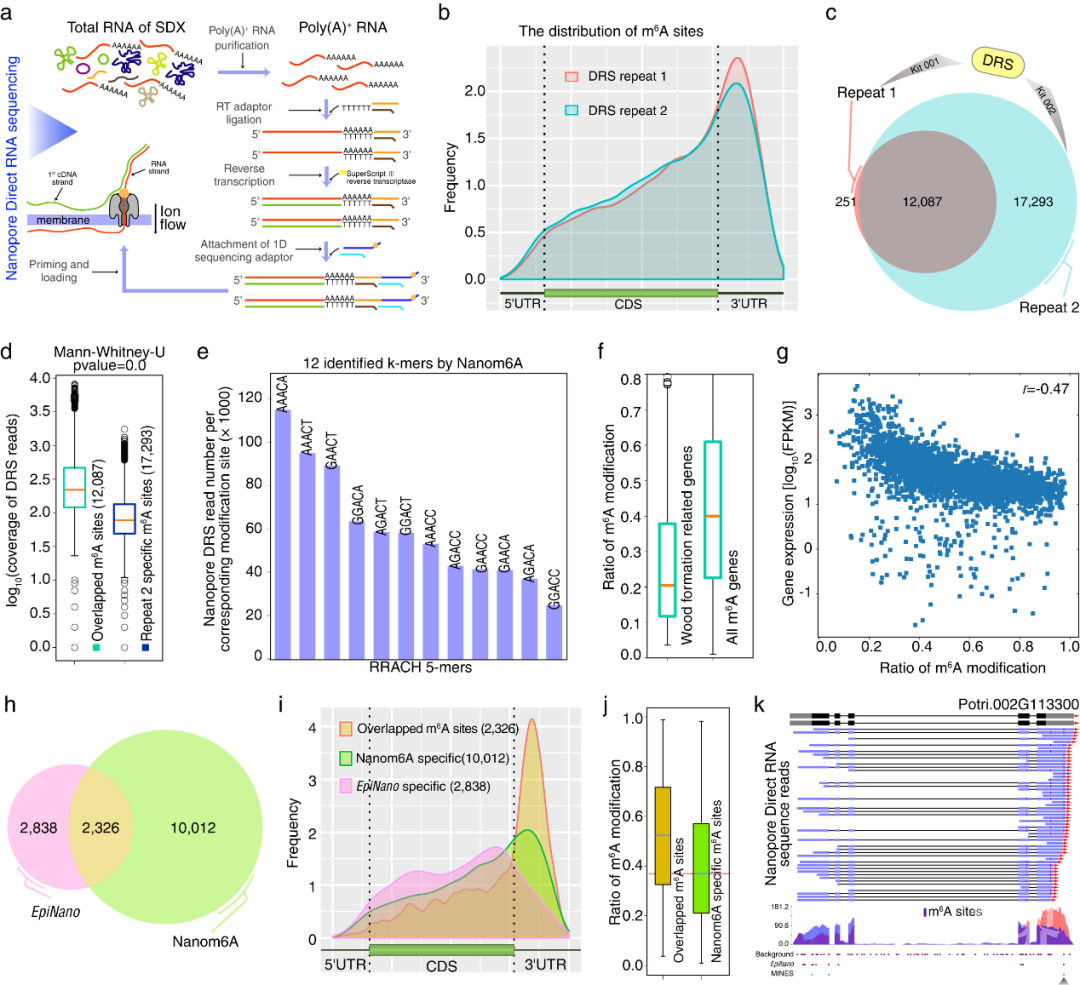
论证了本文方法在动物植物研究中的适用性之后,以毛果杨次生木质部为材料,抽提poly(A)+ RNA进行Nanopore直接RNA测序。检测到在毛果杨次生木质部中m6A位点也富集在转录本的终止密码子或3’UTR上。基因的表达水平和m6A修饰水平呈负相关。与木材合成相关的基因修饰率低,可能与维持这些基因在次生木质部的高表达相关。应用毛果杨次生木质部DRS数据,和另外两种只能定性分析m6A的软件(EpiNano及MINES)进行m6A位点识别比较,发现本文的方法能预测到另外两种算法无法检测到的低修饰率的m6A位点,这些位点大多富集于终止密码子和3’UTR区域。更为重要的是本文的方法除了可以定性识别,还可以进行m6A修饰定量分析,这是EpiNano及MINES所没有的功能。
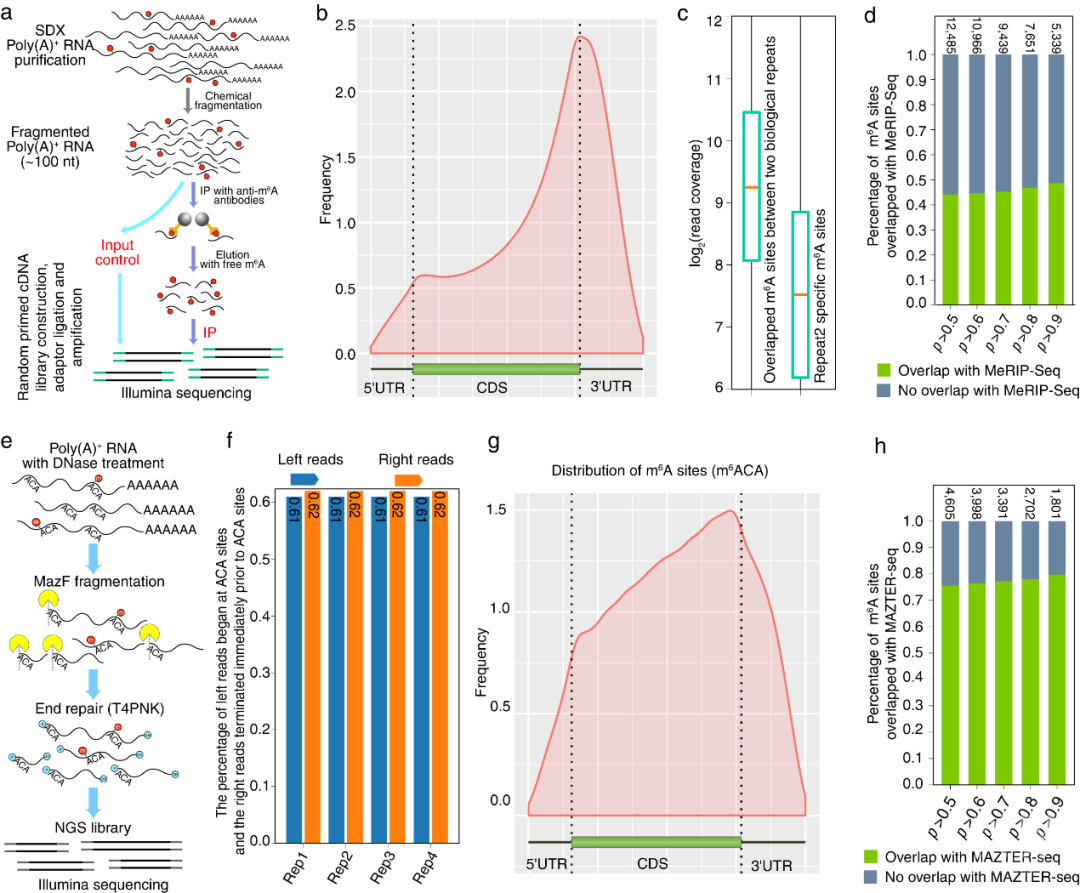
为了验证毛果杨次生木质部中基于DRS数据获得修饰的准确性,进一步运用MeRIP-Seq和m6A-REF-seq两种完全不同的技术,在MeRIP-Seq数据中,片段富集于终止密码子和3’UTR区域。DRS中81%的m6A修饰基因在MeRIP-Seq中被检测到,49%的基于DRS的m6A位点与MeRIP-Seq峰值重叠。在m6A-REF-seq结果中,m6ACA基序同样富集于终止密码子区域,更高精度的m6A-REF-seq有80%的m6A位点与基于DRS的m6A位点重叠。这与m6A-REF-seq (Zhang et al., 2019) 能做到单碱基水平相一致。
最后,运用Nanom6A对毛果杨次生木质部具有选择性多聚腺苷酸化位点的远端和近端poly(A)转录本进行m6A定量分析,发现远端poly(A)和近端poly(A)的m6A修饰比率不同。例如,基因Potri.019G083300,远端poly(A)转录本的m6A修饰比率远少于近端poly(A)转录本。该研究为m6A修饰与选择性多聚腺苷化之间的调控关系提供了重要的线索。
福建农林大学海峡联合研究院林学中心博士生高宇帮和刘旭庆为论文共同第一作者,海峡联合研究院顾连峰教授为通讯作者。林学中心工程师吴碧致、博士生王慧慧、席飞虎、Markus V. Kohnen副教授以及科罗拉多州立大学Anireddy S. N. Reddy教授也参与了本项目。东北林业大学李伟教授课题组在实验材料上提供大量的帮助,中山大学骆观正教授课题组在m6A-REF-seq文库构建上给予了详细的技术指导。该研究得到了国家重点研发基金(2016YFD0600106)的支持。
来源:bioartplants BioArt植物
原文链接:http://mp.weixin.qq.com/s?__biz=MzU3ODY3MDM0NA==&mid=2247501755&idx=1&sn=f92ca4bd9f3e52ceb2d91f5101bd9ec5
版权声明:除非特别注明,本站所载内容来源于互联网、微信公众号等公开渠道,不代表本站观点,仅供参考、交流、公益传播之目的。转载的稿件版权归原作者或机构所有,如有侵权,请联系删除。
电话:(010)86409582
邮箱:kejie@scimall.org.cn



