来源:BioArt
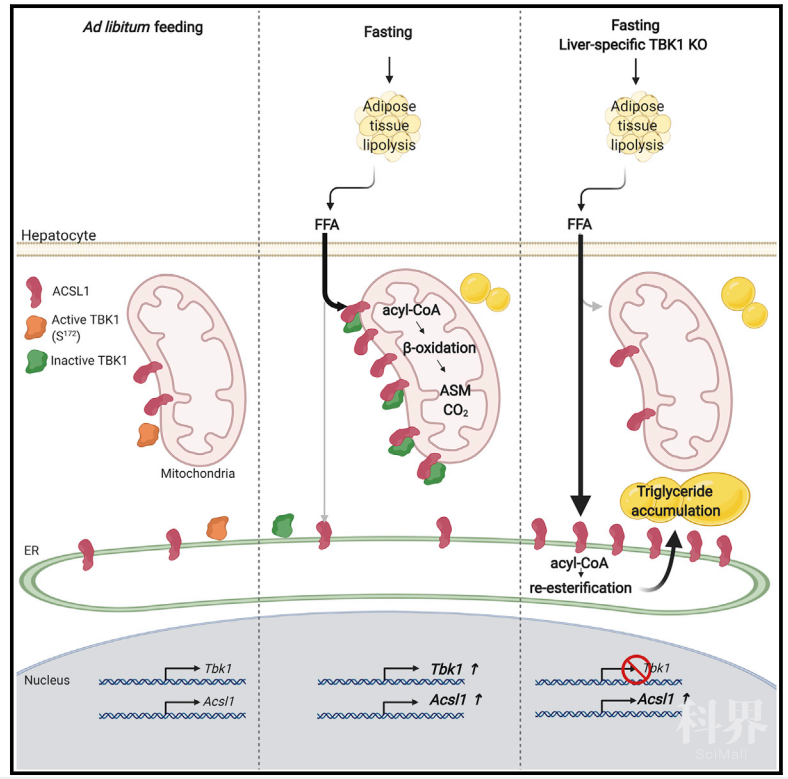
非酒精性脂肪肝(Nonalcoholic fatty liver disease,NAFLD)是由于肝细胞内脂肪堆积过多而引起的病变,常作为肥胖导致的并发症,与II型糖尿病、血脂异常和高血压等密切相关,NAFLD严重时会诱发非酒精性脂肪肝炎 (nonalcoholic Steatohepatitis, NASH) 甚至肝硬化,严重威胁人类健康【1】。据统计,我国成人脂肪肝的患病率高达12.5-35.4%,发病率已经排到了肝病的第一位。肝脏是高等动物脂质代谢的核心器官,肝脏脂肪水平主要取决于游离脂肪酸的摄取和脂类从头生成,而脂肪酸氧化和极低密度脂蛋白的分泌会导致肝脏内脂肪含量降低。长链脂酰辅酶a合成酶ACSL1定位于线粒体和内质网中,是脂肪酸氧化的关键酶之一,ACSL1会在线粒体外膜催化脂酰辅酶a的生成,随后脂酰辅酶a会被CPT1转运到线粒体基质中完成脂肪酸氧化过程,产生乙酰辅酶a并参与各种生命活动【2】。当脂肪酸氧化和脂质分泌水平低于脂质摄取和合成时,NAFLD就会发生。虽然近年来有关NAFLD的基础与临床研究日益受到重视,但脂质稳态、炎症和肝脏病变之间的潜在联系及调控机制仍不清楚,NAFLD的治疗方案也亟待完善。越来越多的研究发现肥胖的发生往往伴随着脂肪组织和肝脏内的轻度炎症反应,且胰岛素抵抗与炎症之间也存在着很强的相关性,炎症反应也是NAFLD向NASH过渡的关键因素【3】。TANK结合激酶1(TANK binding kinase 1, TBK1)是一种丝氨酸/苏氨酸蛋白,在肿瘤发生、免疫应答和自噬等生命活动中具有关键作用,也是脂肪细胞和肝细胞炎症信号传导的效应因子,肥胖会导致肝脏中TBK1的活性上升【4, 5】。临床试验表明,使用TBK1抑制剂氨来占诺(Amlexanox,一种抗炎抗过敏免疫调节剂)治疗后,受试者体内的肝脏脂肪含量能够降低约20%,然而,TBK1调节肝脏能量代谢的机制尚不清楚。近日,加州大学圣地亚哥分校糖尿病与代谢健康研究所所长Alan R. Saltiel教授团队在Cell Metabolism杂志发表了题为TANK-Binding Kinase 1 Regulates the Localization of Acyl-CoA Synthetase ACSL1 to Control Hepatic Fatty-Acid Oxidation 的研究长文,发现TBK1能通过与脂肪酸氧化过程中的关键酶ACSL1的结合影响ACSL1的线粒体定位及肝细胞的脂肪酸氧化,并能通过感知营养状况决定肝细胞中脂肪酸的命运,揭示了TBK1调控肝脏内脂肪代谢的新机制。Alan R. Saltiel教授团队在炎症反应与代谢稳态的互作调控领域深耕多年,并通过一系列原创性工作揭示了NFκB信号通路中的非典型性IκB 激酶TBK1是肥胖、胰岛素抵抗和脂肪细胞能量消耗减少之间的“联系纽带”【4-7】。然而,TBK1在肝细胞中的功能仍知之甚少。为确定TBK1在肝细胞脂肪代谢中的功能,作者首先分析了TBK1在肝细胞中的表达情况,发现禁食处理会诱导肝细胞内TBK1的mRNA和蛋白表达上调,而TBK1的激酶活性在禁食后会降低。高脂膳食(high-fat diet,HFD)处理小鼠后,TBK1的mRNA表达水平与正常饲喂小鼠差异不大,但肝脏内TBK1的酶活性上升。肿瘤坏死因子 (Tumor necrosis factor alpha, TNFα) 具有促炎作用,TNFα处理后脂肪组织中TBK1的表达会发生上调。然而,TNFα处理原代肝细胞后,细胞内一系列炎症反应相关基因的表达虽然会发生上调,但TBK1的表达未发生变化,其激酶活性上升。因此,禁食处理会使肝脏内TBK1表达水平上升,激酶活性下降,而高脂膳食处理能够诱导TBK1的激酶活性。作者在之前研究中发现TBK1抑制剂氨来占诺处理能缓解NAFLD、肥胖及糖尿病等导致的病理反应【8】。在此作者探究了氨来占诺对肥胖小鼠的影响,发现氨来占诺处理8天后肥胖小鼠体重和血糖显著降低,肝脏重量和肝脏脂肪堆积急剧下降;另一方面,氨来占诺处理会导致TBK1活性降低,与脂类合成有关的基因表达水平下调。氨来占诺处理NASH小鼠同样会改善其肝脏代谢状态。因此,抑制TBK1的激酶活性会缓解NAFLD和NASH有关的病理反应。利用肝细胞特异性TBK1敲除小鼠(LTKO)探究TBK1在肝脏代谢中的功能后,作者发现,与外源性氨来占诺处理小鼠不同,肝脏特异性TBK1缺陷会在不诱发炎症反应的前提下促进小鼠肝脏脂肪积累, 并会导致HFD诱导的肝脂质沉着加重,且在禁食后尤为明显。在正常的禁食和喂养周期下,机体能通过平衡合成代谢和分解代谢来维持肝脏脂质水平。为确定TBK1缺陷诱导脂质积累的机制,作者评估了TBK1缺陷小鼠的合成代谢和分解代谢,发现正常或HFD饲喂并不会导致TBK1缺陷小鼠中与脂类代谢有关的基因表达水平改变,也不会造成TBK1缺陷小鼠极低密度脂蛋白分泌、脂类合成及脂肪酸摄取等发生明显变化。然而,TBK1缺陷会降低HFD饲喂后禁食处理小鼠的脂肪酸氧化水平,TBK1是禁食状态下脂肪酸分解代谢所必需的,肝脏能够通过诱导TBK1表达来调节脂肪酸氧化。由于线粒体是脂肪酸氧化的主要场所,因此作者研究了肝脏内TBK1缺陷是否会导致线粒体数量或功能异常。虽然正常或HFD饲喂后LTKO小鼠中一系列与线粒体生物合成有关的基因表达未发生显著变化,但禁食处理后LTKO小鼠肝细胞线粒体体积变大,数量增多。尽管线粒体数量增加,但脂肪酸氧化受到抑制,说明线粒体数量的差异并不能解释LTKO小鼠肝脏脂质过度积累这一表型。进一步分析TBK1缺陷导致肝脏脂肪酸氧化受抑制的机制后,作者发现LTKO小鼠禁食处理后线粒体功能正常,且脂肪酸氧化限速酶CPT1及下游脂肪酸氧化通路并未发生异常变化,但长链脂酰辅酶a合成酶ACSL1的活性降低,从而导致脂酰辅酶A合成发生障碍。分析TBK1在肝细胞线粒体中的蛋白互作关系后,作者发现TBK1与ACSL1存在互作,且TBK1能通过诱导ACSL1的活性而调节长链脂酰辅酶a的合成,进而促进线粒体中的脂肪酸氧化。那么,TBK1是如何调节ACSL1的活性的呢?一方面,作者发现无论正常小鼠还是LTKO小鼠,禁食处理后肝细胞内ACSL1的活性均会上升,证明TBK1并不能直接调节ACSL1的表达和活性;另一方面,TBK1又能够与ACSL1形成复合体,调节脂肪酸氧化。据此,作者推测TBK1对ACSL1活性的调节很可能是通过影响ACSL1的亚细胞定位实现的。分离禁食的正常小鼠与LTKO小鼠肝细胞的各亚细胞组分,并检测TBK1与ACSL1的蛋白表达水平后,作者发现禁食能够促进ACSL1在线粒体中的表达和定位,且TBK1能作为支架蛋白促进ACSL1重定位到线粒体外膜,促进线粒体中的脂肪酸氧化反应。由于TBK1抑制剂处理与TBK1敲除的作用相反,作者接下来探究了TBK1的支架功能是否受到TBK1磷酸化状态(即TBK1活性)的影响。首先,作者利用Co-IP发现ACSL1能与正常TBK1及激酶活性失活的TBK1突变体(K38A)结合,且ACSL1与失活TBK1的结合能力更强,这也证明了ACSL1与TBK1的结合并不依赖于TBK1的催化活性,而TBK1的酶活性甚至抑制了其与ACSL1的结合。其次,通过分析TBK1与ACSL1结合的结构域,作者发现TBK1是通过激酶结构域与ACSL1结合的,该结合作用受到自身磷酸化的负调控。第三,TBK1磷酸化很可能为ACSL1的结合提供了负调控信号,禁食状态下TBK1磷酸化水平较低,ACSL1与TBK1的结合能力增强;而肥胖状态下TBK1磷酸化上调,ACSL1与TBK1的结合能力减弱,导致ACSL1向线粒体的转位受到抑制。论文模式图综上所述,本研究揭示了TBK1能够作为支架蛋白与ACSL1结合,并通过影响ACSL1的线粒体定位,调控了肝细胞中的脂肪酸氧化过程。禁食会导致肝脏内TBK1活性降低,但会提高TBK1作为支架蛋白与ACSL1的结合能力并促进ACSL1定位到线粒体外膜上,从而诱导脂肪酸氧化。而在肥胖状态下,TBK1的活性上升,与ACSL1的结合能力变弱,抑制了ACSL1的线粒体重定位及脂肪酸氧化,诱导了脂肪肝的发生。本研究为TBK1抑制剂在脂肪肝治疗中的应用提供了理论和机理性证据。参考文献:
1. Anstee, Q.M., Targher, G., and Day, C.P. (2013). Progression of NAFLD to diabetes mellitus, cardiovascular disease or cirrhosis. Nat. Rev. Gastroenterol. Hepatol. 10, 330–344.
2. Coleman, R.A., Lewin, T.M., and Muoio, D.M. (2000). Physiological and nutritional regulation of enzymes of triacylglycerol synthesis. Annu. Rev. Nutr. 20, 77–103.
3. Saltiel, A.R., and Olefsky, J.M. (2017). Inflammatory mechanisms linking obesity and metabolic disease. J. Clin. Invest. 127, 1–4.
4. Zhao, P., Wong, K.I., Sun, X., Reilly, S.M., Uhm, M., Liao, Z., Skorobogatko, Y., and Saltiel, A.R. (2018). TBK1 at the crossroads of inflammation and energy homeostasis in adipose tissue. Cell 172, 731–743.e12.
5. Reilly, S.M., Chiang, S.H., Decker, S.J., Chang, L., Uhm, M., Larsen, M.J., Rubin, J.R., Mowers, J., White, N.M., Hochberg, I., et al. (2013). An inhibitor of the protein kinases TBK1 and IKK- 3 improves obesity-related metabolic dysfunctions in mice. Nat. Med. 19, 313–321.
6. Mowers, J., Uhm, M., Reilly, S.M., Simon, J., Leto, D., Chiang, S.H., Chang, L., and Saltiel, A.R. (2013). Inflammation produces catecholamine resistance in obesity via activation of PDE3B by the protein kinases IKKε and TBK1. eLife 2, e01119.
7. Zhao, W. (2013). Negative regulation of TBK1-mediated antiviral immunity. FEBS Lett 587, 542–548.
8. Oral, E.A., Reilly, S.M., Gomez, A.V., Meral, R., Butz, L., Ajluni, N., Chenevert, T.L., Korytnaya, E., Neidert, A.H., Hench, R., et al. (2017). Inhibition of IKK 3 and TBK1 improves glucose control in a subset of patients with type 2 diabetes. Cell Metab 26, 157–170.e7.
来源:BioGossip BioArt
原文链接:http://mp.weixin.qq.com/s?__biz=MzA3MzQyNjY1MQ==&mid=2652508871&idx=5&sn=cfb05f5ecb26c0a687eba12a7b529bf8
版权声明:除非特别注明,本站所载内容来源于互联网、微信公众号等公开渠道,不代表本站观点,仅供参考、交流、公益传播之目的。转载的稿件版权归原作者或机构所有,如有侵权,请联系删除。
电话:(010)86409582
邮箱:kejie@scimall.org.cn



