来源:BioArt
转录因子可以识别特定的基因组序列,以调节复杂的基因表达程序。尽管已经公认转录因子通过碱基读取和形状识别结合到特定的DNA序列上,但对于蛋白质与DNA结合的基础特征仍知之甚少。许多DNA结合蛋白可以诱导DNA结构发生扭转,扭转DNA所需的能量必须来自在复合物形成时发生的分子间相互作用。这种能量损耗可能随序列而变化,且有助于蛋白质-DNA的结合亲和力和选择性。然而通过实验评估能量损耗是具有挑战性的,因为需要精确测量这些扭转DNA构象的丰度,通常情况下是以低丰度的形式存在,因此研究与DNA扭转相关的能量损耗如何有助于蛋白与DNA的识别仍是一个棘手的问题。
2020年10月21日,来自美国杜克大学的Hashim M. Al-Hashimi团队和Raluca Gordân团队合作在Nature杂志上发表了一篇题为“DNA mismatches reveal conformational penalties in protein–DNA recognition” 的文章,在这项研究中,作者开发了一种“SaMBA(saturation mismatch-binding assay)”的高通量测定方法以研究“构象损耗(原文标注为conformational penalty)”在转录因子与DNA识别中的作用。

碱基对错配可以引起DNA发生扭转,其扭转程度显著大于裸露的Watson-Crick序列,例如嘌呤-嘌呤错配(例如G-G和G-A)加宽碱基对,以及嘧啶-嘧啶错配(例如C-T和T-T)会压缩碱基对等。错配使DNA双链不稳定的程度(3.5-10 kBT)与蛋白质结合时扭转DNA的能量损耗相当(3-8 kBT)。作者认为,不同类型的错配可以以多种方式重新分布未结合的DNA,并在某些情况下导致被转录因子(transcription factor, TF)识别的扭转DNA状态的增加。当然,如果通过“预付”一些使DNA扭转的“能量成本”,错配可以反过来增加TF-DNA的结合亲和力。
为深入了解DNA构象损耗在蛋白质-DNA识别中的作用,作者开发了SaMBA,通过在高密度DNA芯片上以高通量方式在已知的TF-DNA结合位点引入每种可能的单碱基改变来产生错配,然后直接在芯片上进行蛋白质结合测量。SaMBA具有很高的重现性,可以使用来自各种独立实验方法的结合测量结果将SaMBA信号强度校准为平衡解离常数(Kd),从而提供了一种以高通量方式确定结合能的途径。当然,除了研究构象损耗在TF-DNA识别中的作用外,SaMBA还可以更广泛地用于检查错配对蛋白质-DNA结合景观的影响以及在突变形成中的作用,包括在某些情况下错配通过产生或增强涉及氢键,静电和堆积的有利相互作用来增强结合。
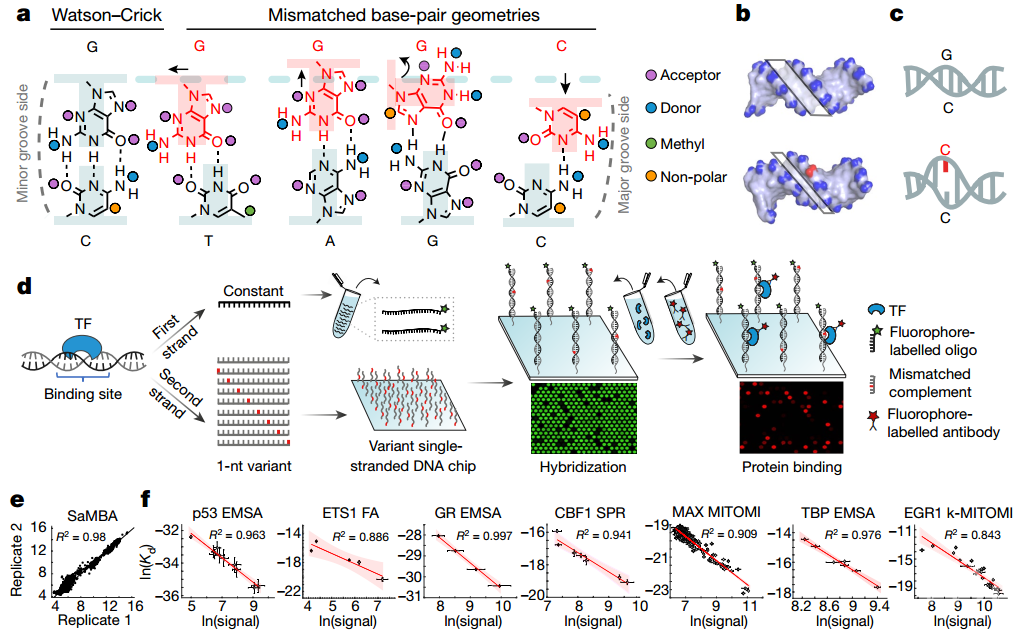
图1,SaMBA可测量错配对高通量蛋白质-DNA结合的影响
随后,作者使用SaMBA获取来自15个不同蛋白家族的22个TFs饱和错配结合图谱,该图谱能够显示由于将每种可能的错配引入已知的TF结合位点及其侧翼区域而引起的蛋白质结合信号的定量变化。尽管在TF结合位点引入的错配的三分之二实质上削弱了结合,但约10%的结合能力增加。甚至在某些情况下,单个错配会形成“超级位点”,与最佳的经典Watson-Crick结合位点相比,其结合亲和力更强。
紧接着,作者使用荧光各向异性(fluorescence anisotropy)和电泳迁移率迁移分析(electrophoretic mobility shift assays, EMSA)验证了错配诱导的TF结合位点增强的代表性实例,并发现相对于Watson-Crick结合位点,结合增加了0.7–2.3 kBT。总体而言,错配引起的对TF结合的影响程度可与Watson-Crick结合位点突变的影响程度相媲美,尽管有时错配与其最接近的突变影响效果相反(例如CG→GG增强结合,而CG→GC减弱结合)。这些数据表明,错配可以提供使用传统高通量方法分析Watson-Crick DNA突变影响所不能涵盖的关于TF-DNA相互作用的更多信息。
对于观察到的错配引起的TF结合亲和力增加的最简单解释是,突变碱基以与错配形状无关的方式与TF形成更有利的相互作用。在这种简单的加性模型中,碱基对中的每个碱基都独立地对TF结合做出贡献,即两个单独的单碱基突变的能量变化总和(增益或损失)等于双突变引起的结合能变化,例如ΔΔGCG> CT +ΔΔGCG> AG =ΔΔGCG > AT(突变的碱基以粗体显示)。作者发现在约42%的错配显着影响TF结合的情况下,可加性在实验误差范围内;然而对于其余约58%的情况,却表现出非加性。这种偏差可能来自多种机制,其中包括错配特异性的改变抵消了与 TF结合的DNA扭转相关的构象损耗而引起的相互作用的增强。对于这种情况,作者期望错配发生在蛋白结合的DNA结构中扭转区域,与料想一致的是,对于RCSB蛋白数据库(Protein Data Bank, PDB)中12个结构可用的TFs子集,发现增强TF结合亲和力的错配结合位点比其余结合位点扭转程度更加明显。
接下来,作者猜测如果错配通过部分“预付”构象损耗而增加结合亲和力,那么错配会模拟与TF结合时DNA的结构。为此,作者使用高分辨率晶体结构数据比较与TF结合的DNA扭转以及错配时的DNA扭转,并在66%的示例中观察到某种形式的结构模仿。例如,ETS1特定位置的GA错配,能够增加2.3 kBT的结合力,且错配时DNA扭转模仿了ETS1与DNA结合时的伸张,C1'–C1'距以及小沟宽度。类似地,如下图所示,p53–DNA复合物的晶体结构在红色标记的位置显示出狭窄的Hoogsteen构象,而C-T和T-T错配会增加p53-DNA的结合亲和力,通过限制C1'-C1'的距离和小沟宽度来模拟Hoogsteen碱基配对。这些数据表明,错配通过有效地模拟蛋白-DNA结合的结构特征,从而先“预付”一些能量损耗以助于形成优选的结合结构。
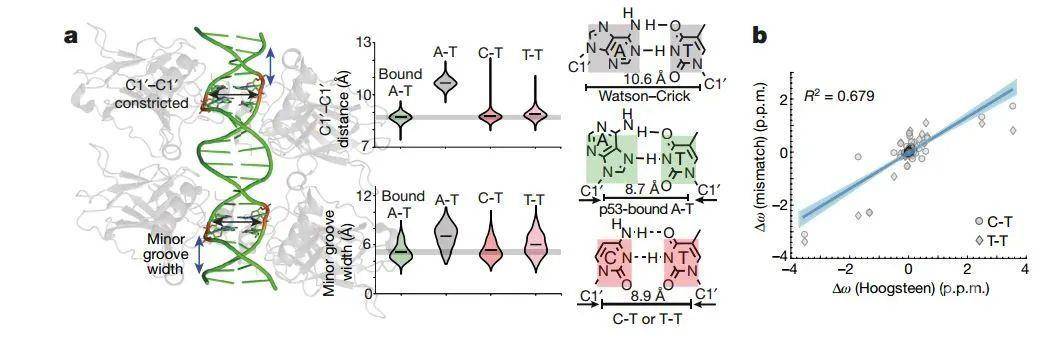
图2,以p53-DNA结合为例,错配会模仿Hoogsteen A-T碱基对的几何学特征
总的来说,这项研究提供了迄今为止最大的关于DNA错配对蛋白质结合影响的分析,并揭示了DNA构象损耗是决定蛋白质-DNA结合亲和力和选择性的重要因素。这项工作提供的检测方法可以扩展到包括由多个错配,插入和缺失以及受损和表观遗传修饰的核苷酸引起的DNA扭转,因此可以用于以高通量和无偏见的方式彻底研究不同类型的能量损耗。此外,无论错配增强TF结合的确切机制如何,这些高亲和力相互作用都可以提供抑制特定错配位点修复的生物物理机制,从而有助于理解细胞中基因突变的形成。
来源:BioGossip BioArt
原文链接:http://mp.weixin.qq.com/s?__biz=MzA3MzQyNjY1MQ==&mid=2652504516&idx=6&sn=85d8eb375c8b809de828d3d508a49155&chksm=84e19270b3961b66bd829a38e78e58244aa9726c9febc3a4ffdf62ffad27f305bcd6c09bd138&scene=27#wechat_redirect
版权声明:除非特别注明,本站所载内容来源于互联网、微信公众号等公开渠道,不代表本站观点,仅供参考、交流、公益传播之目的。转载的稿件版权归原作者或机构所有,如有侵权,请联系删除。
电话:(010)86409582
邮箱:kejie@scimall.org.cn

