来源:生物通
波士顿大学口腔医学院的Andrea Geisz和Miklós Sahin-Tóth开发出一种T7D23A基因敲入小鼠,以填补此领域的空白。这种小鼠携带阳离子胰蛋白酶原(T7亚型)的杂合p.D23A突变,为胰腺炎的研究和治疗提供了出色的模型。
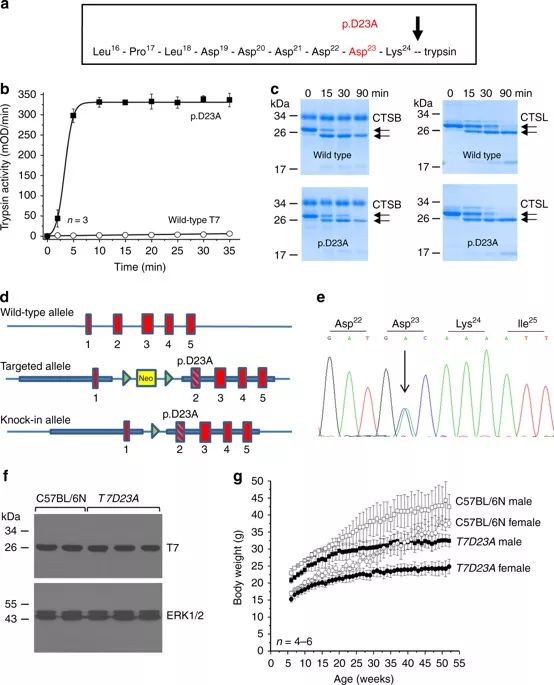
图1. p.D23A突变对胰蛋白酶原T7亚型的影响
胰腺炎是因为胰蛋白酶的自身消化作用而引起的疾病,包括急性胰腺炎、复发性急性胰腺炎和慢性胰腺炎。这些疾病常常引起腹痛、恶心、呕吐、发热等症状,但并没有特异性疗法。
以往的研究表明,编码消化蛋白酶或其抑制剂的基因若发生突变,则往往会促进急性胰腺炎的发生,并逐渐发展成慢性胰腺炎。例如,PRSS1(阳离子胰蛋白酶原)、CTRC(胰凝乳蛋白酶C)以及SPINK1(Kazal型丝氨酸蛋白酶抑制剂1)的致病突变通过刺激自发激活或干扰保护机制,从而促进胰蛋白酶原转化成有害的胰蛋白酶。
不过,基因突变引起的胰腺内胰蛋白酶原自发激活具有直接致病作用,这虽然获得了大量的遗传和生化证据支持,但目前还缺乏适当动物模型的确认。更重要的是,动物模型的缺乏还阻碍了胰蛋白酶抑制剂的临床前试验。
为此,波士顿大学口腔医学院(Henry M. Goldman School of Dental Medicine)的Andrea Geisz和Miklós Sahin-Tóth开发出一种T7D23A基因敲入小鼠,以填补此领域的空白。这种小鼠携带阳离子胰蛋白酶原(T7亚型)的杂合p.D23A突变,为胰腺炎的研究和治疗提供了出色的模型。这项成果近日发表在《Nature Communications》上。
T7D23A基因敲入小鼠的开发
小鼠基因组包含20个胰蛋白酶原基因。之前的研究表明,只有四种胰蛋白酶原高水平表达,它们分别是:T7、T8、T9和T209亚型。其中,T7亚型占胰蛋白酶原总量的40-50%,并且与其他亚型相比自发激活更快,产生的胰蛋白酶水平也更高。
为了产生自发激活增加的T7突变体,研究人员在激活肽中引入了Ala突变,以取代Asp23(p.D23A)。为了在体外测试T7 p.D23A突变体的激活特性,他们纯化了重组野生型和突变型T7胰蛋白酶原,并测定了自发激活。与野生型相比,p.D23A突变体的自发激活显著增加了50倍,同时,其他胰蛋白酶激活和降解途径不受影响。
之后,研究人员委托赛业生物(Cyagen)制备了T7D23A基因敲入小鼠,具体是通过C57BL/6胚胎干细胞中的同源重组将p.D23A突变引入小鼠基因组(图1)。DNA测序表明,这些杂合小鼠在突变位点显示出两个高度相当的峰,表明野生型和突变型等位基因的表达水平相当。新的T7D23A品系未出现明显的表型改变,并且正常繁殖。
T7D23A小鼠出现自发的急性和慢性胰腺炎
研究人员对T7D23A小鼠的胰腺形态进行观察,发现2-3周时是正常的,但4-5周时变化较大,而2个月后胰腺明显变小。组织学检测发现,T7D23A小鼠从第3周开始出现典型的急性胰腺炎,在4-5周出现得更加频繁(~40%)。水肿破坏了紧密排列的组织结构,大量炎性细胞浸润,且小叶出现中心局部坏死(图2)。
在4-5周时,研究人员发现超过一半的小鼠表现出明显的疾病进展。它们的胰腺切片显示出广泛的再生指征,包括假管复合物出现、胰管扩张和弥漫性间质纤维化,这些都是早期慢性胰腺炎的特征。从2个月开始并持续到6至12个月,脂肪开始在组织学图像中占据主导地位,而炎性细胞和假管复合物的数量开始减少。在扩张胰管的周围纤维化更普遍,有时还充满嗜酸性物质,这些是晚期慢性胰腺炎的特征。
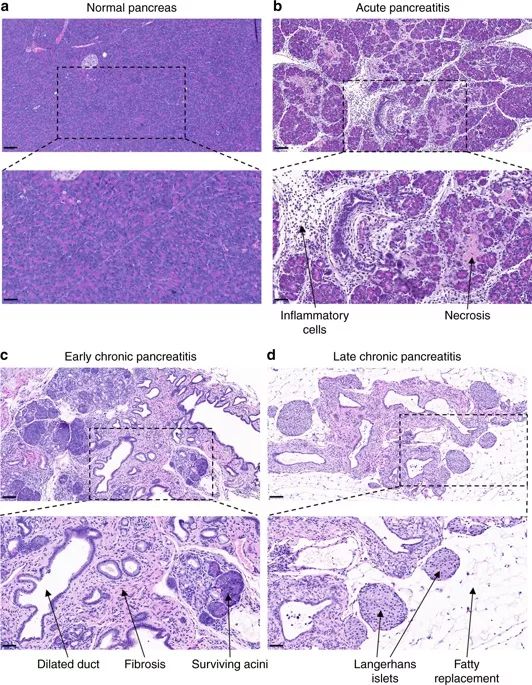
图2. T7D23A小鼠的胰腺组织学
之后,研究人员将T7D23A小鼠的胰腺重量与C57BL/6N对照小鼠进行比较,发现约40%的小鼠在4-5周龄表现出明显增加。这种增加与组织切片上观察到的水肿性急性胰腺炎一致。大约50%的小鼠在4-5周龄时胰腺重量明显减轻,表明早期慢性胰腺炎的出现。所有T7D23A小鼠在2月、6月和12月龄时显示出与晚期慢性胰腺炎相关的胰腺萎缩,与对照小鼠相比胰腺重量减轻约70%。
T7D23A小鼠的酶活检测
淀粉酶是诊断急性胰腺炎最常用的指标。因此,研究人员也测定了各个年龄的T7D23A小鼠血浆中的淀粉酶活性。在4-5周时,他们在约40%的小鼠中观察到淀粉酶值偏高,这与之前的结果一致。不过,并非所有小鼠都能捕获到急性胰腺炎的指标,这可能与采样时间和发病年龄有关,也可能是小鼠在没有急性发作的情况下患上慢性胰腺炎。
接着,他们以3周龄(无病理变化)、4周龄(急性或早期慢性胰腺炎)和2月龄(晚期慢性胰腺炎)T7D23A小鼠为对象,测定了新鲜制备的胰腺匀浆液中的胰蛋白酶活性。与相同年龄的对照小鼠相比,3周时未检测到胰蛋白酶活性的升高,但在4周时检测到高值。尽管功能性腺泡细胞减少,但在2个月时仍观察到酶活的升高,表明胰腺内胰蛋白酶的激活在整个疾病过程中持续存在。这些结果支持了,胰蛋白酶原的自发激活可引起急性胰腺炎发作,且随后发展成慢性胰腺炎。
为了排除与胰蛋白酶原自发激活无关的疾病机制,研究人员还制备了p.D23A、K24G双突变的小鼠品系,除了带有杂合的T7D23A等位基因,胰蛋白酶原激活位点Lys24也经过突变。这种小鼠在5.5个月时没有出现自发的胰腺病理变化。这个结果提供了令人信服的证据,说明胰蛋白酶原错误折叠等机制并不引起T7D23A小鼠的表型。
T7D23A小鼠与人类胰腺炎的特征比较
研究人员认为,这种新模型概括了人类胰腺炎的临床疾病特点,它总是先发展为急性胰腺炎,然后再发展为不可逆的慢性胰腺炎。不过,他们没有观察到人类经常出现的急性病情反复发作。这种差异可能是由此处所用的胰蛋白酶原突变造成的。
研究人员认为,他们展示了第一个慢性胰腺炎的临床前小鼠模型,该疾病是由胰腺内胰蛋白酶原自发激活的增加所驱动的。这个模型概括了人类慢性胰腺炎的显著特征。值得一提的是,尽管T7D23A小鼠出现明显的病理变化,但它们能够正常繁殖并发育到成年期,这是临床前应用的先决条件。
“在概念层面,这个模型提供了直接的体内证据,表明胰蛋白酶原的自发激活可驱动慢性胰腺炎的发作和发展,且治疗方法应针对胰腺内的胰蛋白酶,”研究人员总结道。
参考文献:
A preclinical model of chronic pancreatitis driven by trypsinogen autoactivation
来源:gh_c1fce5726992 生物通
原文链接:https://mp.weixin.qq.com/s?__biz=MjM5NzMwNjYyMg==&mid=2675530667&idx=2&sn=f0b2af4b1a4798b54e6d3d72ccfeb282&chksm=bc51f23a8b267b2cbba8289c5a94862745afe1d225ba80c53db8662a829c15210e54defcf932&scene=27#wechat_redirect
版权声明:除非特别注明,本站所载内容来源于互联网、微信公众号等公开渠道,不代表本站观点,仅供参考、交流、公益传播之目的。转载的稿件版权归原作者或机构所有,如有侵权,请联系删除。
电话:(010)86409582
邮箱:kejie@scimall.org.cn



