来源:BioArt
肺纤维化既是独立的疾病也是多种慢性纤维增生性肺病的基本病理改变,是许多慢性肺病致残、致死的重要原因【1】。但是,肺纤维化的发病机制尚未完全阐明,更重要的是,至今也没有公认的、真正能够逆转肺纤维化的药物。虽然吡非尼酮和尼达尼布已被FDA批准用于治疗特发性肺纤维化,但是这些药物仅能延缓肺纤维化的发病,并不能逆转纤维化的病理改变【2】。深入研究肺纤维化的发病机制,发现并鉴定新的药靶、开发有效防治肺纤维化的药物是医药科研人员面对的紧迫挑战。
2019年8月27日,中国医学科学院药物研究所天然药物活性物质与功能国家重点实验室胡卓伟研究员团队在肺纤维化发病的分子机制研究方面取得重要进展,该研究以Targeting the Transcriptional Factor C/EBPβ Degradation Reduces Lung Fibrosis by Restoring Activity of the Ubiquitin-Editing Enzyme A20 in Macrophages为题发表在Immunity上。该研究揭示蛋白激酶GSK-3β通过与泛素编辑酶A20相互作用,抑制其酶活性,导致其底物蛋白核转录因子C/EBPβ大量堆积在巨噬细胞,引起并维持巨噬细胞促纤维化表型,从而促进肺纤维化的发生和发展。这一发现为肺纤维化的治疗提供了新的药物靶点。

近年来,胡卓伟研究团队致力于从免疫生物学角度研究肺纤维化发病的分子细胞生物学机制,在此基础上研发能够预防和治疗肺纤维化的药物。巨噬细胞作为先天免疫细胞,可分泌多种炎性介质参与肺纤维化的发病【3,4】。泛素编辑酶A20通过调节多种细胞生物学功能参与炎性疾病过程【5】。但A20是否调节巨噬细胞功能参与肺纤维化发病仍不清楚。
本研究中该团队发现在巨噬细胞中过表达A20显著抑制博来霉素所致的小鼠肺纤维化,而在巨噬细胞中特异性敲除A20则显著加重博来霉素所致的小鼠肺纤维化。随后,该研究团队利用单细胞测序方法分析了过表达A20对巨噬细胞表型的影响。结果显示,过表达A20显著抑制了肺泡巨噬细胞的促纤维化表型(图1)。重要地是,作者发现,肺纤维化病人和动物的肺泡巨噬细胞内A20的酶活性受到了显著抑制,引起并维持了肺泡巨噬细胞的促纤维化表型。
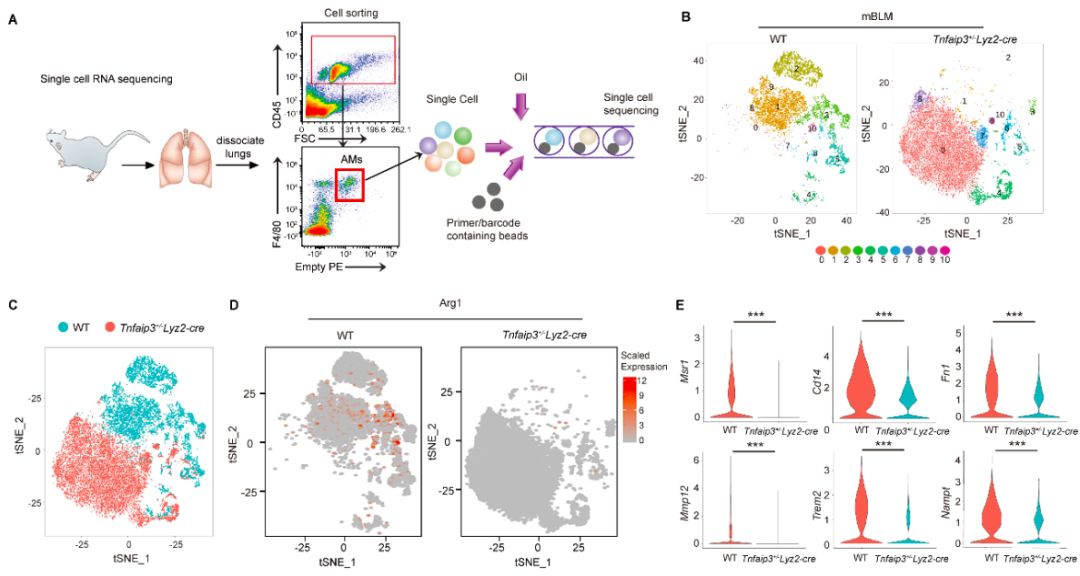
图1. 肺泡巨噬细胞表型决定肺纤维化发生发展。单细胞测序结果显示,肺泡巨噬细胞过表达A20使肺泡巨噬细胞由促纤维化表型转变为抗纤维化表型。
进一步的机制研究发现,巨噬细胞中转录因子C/EBPβ是A20的泛素化修饰底物,A20通过去掉K63泛素化修饰链,增加K48泛素化修饰链,促进C/EBPβ的降解,而肺纤维化时A20活性的抑制导致肺泡巨噬细胞中C/EBPβ大量堆积,促进肺泡巨噬细胞向促纤维化表型的转化。该团队还发现蛋白激酶GSK3β在肺纤维化组织中表达增加,高表达的GSK3β导致A20的磷酸化,抑制A20的酶活性,促进巨噬细胞的活化(图2)。这些结果提示GSK3β-A20-C/EBPβ轴是导致肺纤维化组织中肺泡巨噬细胞活化的重要原因,而GSK3β/A20相互作用是治疗肺纤维化的重要靶点。在此基础上,该团队研究人员通过分析GSK3β与A20相互作用结构域氨基酸序列,筛选到可以抑制GSK3β和A20结合的小分子多肽探针。随后该研究团队证实该干扰肽在不同肺纤维化模型中均产生很好的治疗效果。
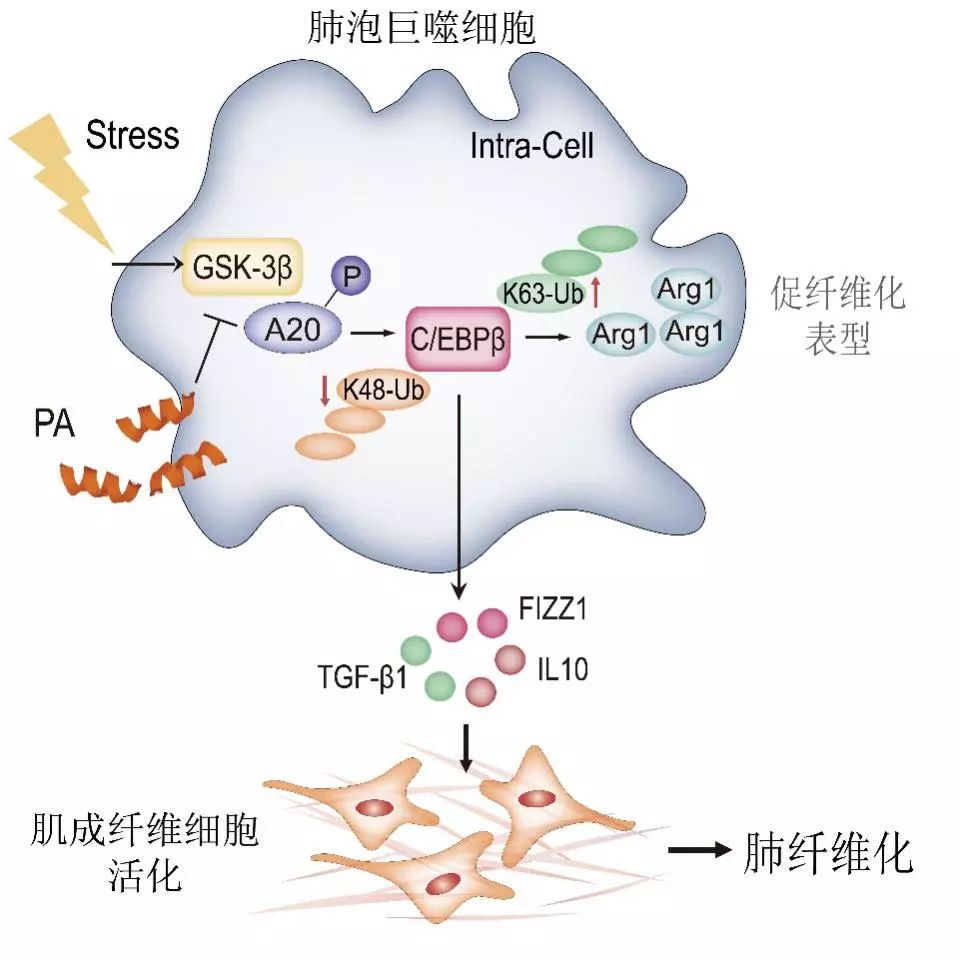
图2. GSK-3β与A20相互作用导致A20酶活性的抑制,引起其底物转录因子C/EBPβ大量堆积在巨噬细胞,因此导致并维持巨噬细胞促纤维化表型,促进肺纤维化发病。
这项研究从全新角度阐释了肺纤维化发病的分子机制,不但鉴定和发现了GSK3β/A20相互作用这一肺纤维化治疗的潜在新靶点,更发现了靶向该相互作用的治疗性化合物,展现了分子机制研究向临床应用转化的潜力。
该论文通讯作者为中国医学科学院药物研究所胡卓伟研究员。助理研究员刘姗姗和吕晓希作为该论文的共同第一作者,对本研究做出了主要贡献。
原文链接:
https://doi.org/10.1016/j.immuni.2019.06.014
参考文献
1. Putman, R.K., Hatabu, H., Araki, T., Gudmundsson, G., Gao, W., Nishino, M., Okajima, Y., Dupuis, J., Latourelle, J.C., Cho, M.H., et al. (2016). Association Between Interstitial Lung Abnormalities and All-Cause Mortality. JAMA 315, 672-681.
2. Richeldi, L., Collard, H.R., and Jones, M.G. (2017). Idiopathic pulmonary fibrosis. Lancet 389, 1941-1952.
3. He, C., Ryan, A.J., Murthy, S., and Carter, A.B. (2013). Accelerated development of pulmonary fibrosis via Cu,Zn-superoxide dismutase-induced alternative activation of macrophages. J Biol Chem 288, 20745-20757.
4. Larson-Casey, J.L., Deshane, J.S., Ryan, A.J., Thannickal, V.J., and Carter, A.B. (2016). Macrophage Akt1 Kinase-Mediated Mitophagy Modulates Apoptosis Resistance and Pulmonary Fibrosis. Immunity 44, 582-596.
5. Ma, A., and Malynn, B.A. (2012). A20: linking a complex regulator of ubiquitylation to immunity and human disease. Nat Rev Immunol 12, 774-785.
来源:BioGossip BioArt
原文链接:http://mp.weixin.qq.com/s?__biz=MzA3MzQyNjY1MQ==&mid=2652474535&idx=3&sn=2864a80ee4a36cd17f56b86a0170e784&chksm=84e21f13b3959605b8001f7b1db4596f7fa84937f95bb997bfaa591d02da250ac21991724b26&scene=27#wechat_redirect
版权声明:除非特别注明,本站所载内容来源于互联网、微信公众号等公开渠道,不代表本站观点,仅供参考、交流、公益传播之目的。转载的稿件版权归原作者或机构所有,如有侵权,请联系删除。
电话:(010)86409582
邮箱:kejie@scimall.org.cn



