来源:植物科学最前沿
工程Cas9变体(SpCas9s)和酸性氨基球菌种Cas12a(AsCas12a)在细胞中的脱靶现象比酿脓链球菌Cas9(SpCas9)少。目前,核酸酶的特异性是根据靶标和脱靶基因组位点的DNA断裂修复疤痕来推断的。此类脱靶检测策略无法将酶内在动力学参数与核酸酶递送方法、暴露时间、遗传背景、细胞周期阶段或DNA断裂修复途径等因素区分开。大多数基于体外下一代测序(NGS)的策略还旨在在基因组中找到假定的脱靶位点,但是它们比较reads数而不是动力学速率,并且无法识别DNA末端或其加工动力学。为了直接测定和预测这些酶的特异性,需要在系统的脱靶DNA序列文库中比较脱靶结合亲和力和切割动力学。最近,德克萨斯大学奥斯汀分校的 Ilya J. Finkelstein 团队在Nature Biotechnology上发表了题名为“Massively parallel kinetic profiling of natural and engineered CRISPR nucleases”的研究论文,主要建立了一个新的实验平台,该平台可以全面测量合成DNA文库中DNA结合和切割特异性去检测CRISPR-Cas核酸酶。

NucleaSeq是一个快速、大规模平行的体外平台,能够测量CRISPR-Cas核酸酶的裂解动力学。NucleaSeq捕获了引导RNA(gRNA)匹配和错配的DNA序列的大型文库的切割产物的时间分辨身份。通过芯片杂交的关联作图平台(CHAMP)在重新设计的NGS MiSeq芯片上测量了这些文库的核酸酶结合特异性。结合NucleaSeq和CHAMP,研究人员评估了五个SpCas9变体和Cas12a的DNA,这些DNA包含gRNA相关的错配、插入和缺失(图1)。工程Cas9s,特别是Cas9-HF1,增加了切割特异性,但没有增加结合特异性。令人惊讶的是,虽然Cas12a在细胞中具有较高的切割特异性,但它在体外具有与wtCas9相似的特异性。最初的DNA切割位点和随后的末端修饰会随核酸酶、gRNA和RNA–DNA错误配对的位置而变化。有趣的是,PAM远端RNA-DNA错配会通过核酸酶末端修饰产生不相容的DNA末端,但不会减慢整体切割速率(图2)。
总的来说,研究人员使用大量数据去开发了一个生物物理学模型,该模型提供了比较CRISPR核酸酶的定量框架,并揭示了脱靶切割的机理见解。更广义的层面上来说,NucleaSeq和CHAMP能够对工程核酸酶和天然核酸酶的特异性和裂解产物进行快速、定量和系统的比较。

图1:整合NucleaSeq和CHAMP的平台概观。
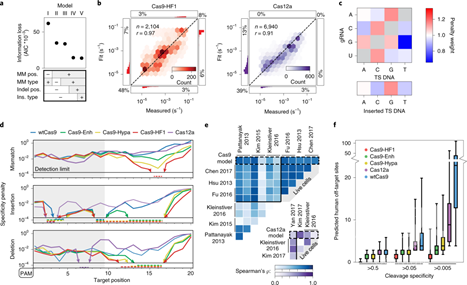
图2:CRISPR-Cas核酸酶切割的统计建模。
来源:frontiersin 植物科学最前沿
原文链接:https://mp.weixin.qq.com/s?__biz=MzIyOTY2NDYyNQ==&mid=2247500292&idx=4&sn=1a5e81d0feb3875226e1e994522019ec&chksm=e8bdb01adfca390cf579a154a79412353667214d3ceae362e44b0013b4eca28c4e094daec65f#rd
版权声明:除非特别注明,本站所载内容来源于互联网、微信公众号等公开渠道,不代表本站观点,仅供参考、交流、公益传播之目的。转载的稿件版权归原作者或机构所有,如有侵权,请联系删除。
电话:(010)86409582
邮箱:kejie@scimall.org.cn

