来源:药学进展
专家介绍:卢娜

教授,博士生导师。中国药科大学基础医学与临床药学学院基础医学系主任,江苏省肿瘤发生与干预重点实验室副主任。研究方向:肿瘤药理学。2014年2月—2015年3月赴美国密歇根大学做访问学者。获2015年度高等学校科学研究优秀成果奖(科学技术)一等奖和2015年度江苏省科学技术奖一等奖。获2012年度“新世纪优秀人才”资助。获2013年度江苏省杰出青年科学基金资助。获2016年度江苏省“333工程第二层次”中青年科技领军人才称号。依托江苏省肿瘤发生与干预重点实验室,进行肿瘤的发病机制及抗肿瘤药物的作用机制研究。近年来以第一作者或通讯作者发表SCI研究论文70余篇。主持国家自然科学基金(2项)、“十三五”重大新药创制专项(1项)、“十二五”重大新药创制专项(1项)、江苏省杰出青年科学基金(1项)、江苏省自然科学基金(1项)等多项国家及省部级项目。获得专利授权2项,申请专利2项。
正文
细胞周期蛋白依赖性激酶9及其抑制剂研究进展
阚少鑫,卢娜
(中国药科大学基础医学与临床药学学院江苏省肿瘤发生与干预重点实验室,江苏 南京 210009)
[摘要]细胞周期蛋白依赖性激酶(CDK)在细胞中不仅负责细胞周期调控,也在细胞转录过程中作为调控因子发挥着重要作用。CDK9作为CDK家族的一员,在细胞转录调控中起重要作用。CDK9和细胞周期蛋白T1结合形成正性转录延长因子b,后者通过磷酸化RNA聚合酶Ⅱ的碳端结构域CTD来调节转录延伸。CDK9抑制剂以竞争性结合的方式,抑制CDK9介导的转录延伸阶段。简要介绍CDK9的功能及其在肿瘤中的作用机制,并总结CDK9抑制剂的研究进展。
细胞周期蛋白依赖性激酶(cyclin-dependent kinase,CDK)是一类丝氨酸/苏氨酸激酶,它们在细胞转录、细胞周期以及神经分化过程中发挥着重要的作用。CDK按照功能可以分为2种:一种控制细胞周期,一种调节细胞转录。CDK9和其他CDK不同的是,它只在转录延长阶段发挥作用,不参与细胞周期的调节。CDK9与细胞周期蛋白(分别是T1、T2和K)形成异源二聚体,即正性转录延长因子b(positive transcription elongation factor b,P-TEFb)。P-TEFb能够磷酸化RNA聚合酶Ⅱ(RNA polymerase II,Pol II)的碳末端结构域上的2位丝氨酸,激活PolⅡ。
近来研究证实了CDK9在许多病理过程中的重要性,如肿瘤、病毒复制、炎症反应及心血管疾病等。
CDK9存在于所有的哺乳动物细胞中,其能与对应的细胞周期蛋白结合形成P-TEFb,促进转录延长。人类免疫缺陷病毒1(human immunodefciency virus 1,HIV1)的转录主要由反式激活蛋白(trans activating protein,TAT)控制。TAT直接与P-TEFb的亚基CycT1结合,而P-TEFb由CDK9和CycT1组成。这些分子对PolⅡ的碳端结构域(carbon terminal domain,CTD)的磷酸化起着重要作用。
中性粒细胞的凋亡以及被巨噬细胞所清除是炎症消散的关键步骤。如果这个过程没有得到控制,将会发生慢性炎症;此外中性粒细胞的过度活化或因坏死而释放出一些毒性小颗粒物质均会引起组织损伤。研究证明,Mcl-1蛋白对中性粒细胞的存活起着重要作用。通过抑制CDK9的活性,可以抑制Mcl-1蛋白的表达,从而可以促进中性粒细胞的凋亡,进而促进炎症消散。
病理性心肌肥大是心肌细胞和冠状动脉血管生长不平衡的结果。心肌细胞肥大可归因于细胞内整体RNA含量升高所导致的蛋白质合成增加,而负责编码RNA转录的PolⅡ被认为是心肌肥大的限制因素。
越来越多的研究表明,CDK9与血液肿瘤、肝癌、前列腺癌、成神经细胞瘤等有关。因此,CDK9被认为是治疗肿瘤的一个潜在的药物靶点。
本文综述了CDK9的功能及CDK9抑制剂的研究进展,旨在为CDK9抑制剂类抗肿瘤药物的研发提供参考。
1CDK9的生物学功能
CDK9最初因其特征性的氨基酸基序(Pro-Ile-Thr-Ala-Leu-Arg-Glu)被命名为PITALRE,其功能首次在人类免疫缺陷病毒的研究中得到阐明。CDK9在细胞中包括CDK942与CDK955这2种亚型。其中,在结构上CDK955仅在N端比CDK942多出117个氨基酸,2种亚型的磷酸化的方式可能相同,但是二者在细胞内分布的位置、表达的模式、调节的方式,以及在组织中的表达都有差异。据报道,CDK942主要在核质中,而CDK955在核仁中。在未分化的单核细胞中,CDK942的表达水平高于CDK955,而在巨噬细胞分化时会诱导CDK955的表达。在原始淋巴细胞中虽然CDK955与CDK942的基因编码相同,但二者的启动子区域不同。所以2种亚型的差异性表达和细胞信号传导以及细胞类型有关。二者在组织中分布也有差异,CDK942主要分布在睾丸和脾,而CDK955主要分布在肝组织、脑和肺。
在细胞中这2种亚型都可以与细胞周期蛋白(分别是T1、T2和K)形成P-TEFb复合物。但由于CDK9的单体不够稳定,所以在与细胞周期蛋白结合前,需要与伴侣蛋白(CDC37、HSP70和HSP90)结合形成瞬时复合物。随后,CDK9从这个瞬时复合物释放出来,再与细胞周期蛋白结合。CDK9与细胞周期蛋白结合以后,CDK9T环上的第186位苏氨酸残基(T186)发生磷酸化,从而活化P-TEFb。活化后的P-TEFb可以磷酸化PolⅡCTD的2位丝氨酸(Ser2),从而使得转录的延伸阶段得以正常进行(如图1所示)。
2CDK9在肿瘤中的作用机制
肿瘤被认为是一种增殖性疾病,肿瘤细胞的生存主要依赖于抗凋亡蛋白。研究表明,通过抑制CDK9介导的转录过程,可以减少抗凋亡蛋白的表达,进而促使肿瘤细胞发生凋亡。
2.1急性髓系白血病
CDK9作为转录过程中的重要角色,可以调控下游Mcl-1、Bcl-2、XIAP和Myc等短寿命凋亡蛋白。通过抑制CDK9的活性,能够降低这些抗凋亡蛋白的水平,从而诱导癌细胞的凋亡,尤其是在人类髓系白血病中,以急性髓系白血病(acute myeloid leukemia,AML)最为敏感。在由核型定义的AML的不同的亚型的数据库中,研究人员可以直接比较某个基因在白血病细胞和正常细胞中的表达水平。研究者通过该数据库发现,在AML样本中CDK9的mRNA表达水平增加,且在AML的不同亚型之间表达也有所差异。
AML占成人急性白血病的80%,患者的主要治疗方法是联合治疗,并且在33%~57%的AML病例中可见多药耐药,所以需要新的分子靶向疗法来更广泛地改善AML的治疗效果和预后。在许多血液肿瘤和实体恶性肿瘤中Myc是过表达的,它受到多个信号级联的调节,并且调节许多细胞过程(包括增殖、细胞周期进程、分化和细胞凋亡)中的转录因子。因此,Myc的失调可能导致不受控制的细胞增殖,基因组不稳定以及逃避免疫监视。尽管Myc在白血病中具有重要作用,但是通过药理学途径直接靶向Myc较为困难。但通过间接途径干扰Myc,可以起到抗AML的疗效。
CDK9介导Mcl-1和Myc的转录,而Mcl-1以及Myc对肿瘤细胞的生长和存活至关重要,抑制CDK9可导致肿瘤细胞发生凋亡。其中CDK9通路对Mcl-1和Myc的调节跟AML发病机制有关。有研究表明,在R/RAML的病例中,有一半都有Mcl-1的表达升高,并且预后不良。在小鼠模型中,Mcl-1的高表达与AML的发生有关,同时Mcl-1对人AML细胞的存活和增殖也起着关键作用。研究者发现混合谱系白血病(mixed lineage leukemia,MLL)基因常与11-19白血病蛋白(eleven-nineteen leukemia,ENL)基因、激活增强子结合蛋白4(activating enhancer binding protein 4,AF4)基因、AF9基因和AF10基因等伙伴基因形成MLL融合基因。研究表明,ENL不仅是PolⅡ的延伸因子,也是延伸阶段促进转录的同源核蛋白,因此MLL-ENL融合蛋白可以影响转录延伸过程。
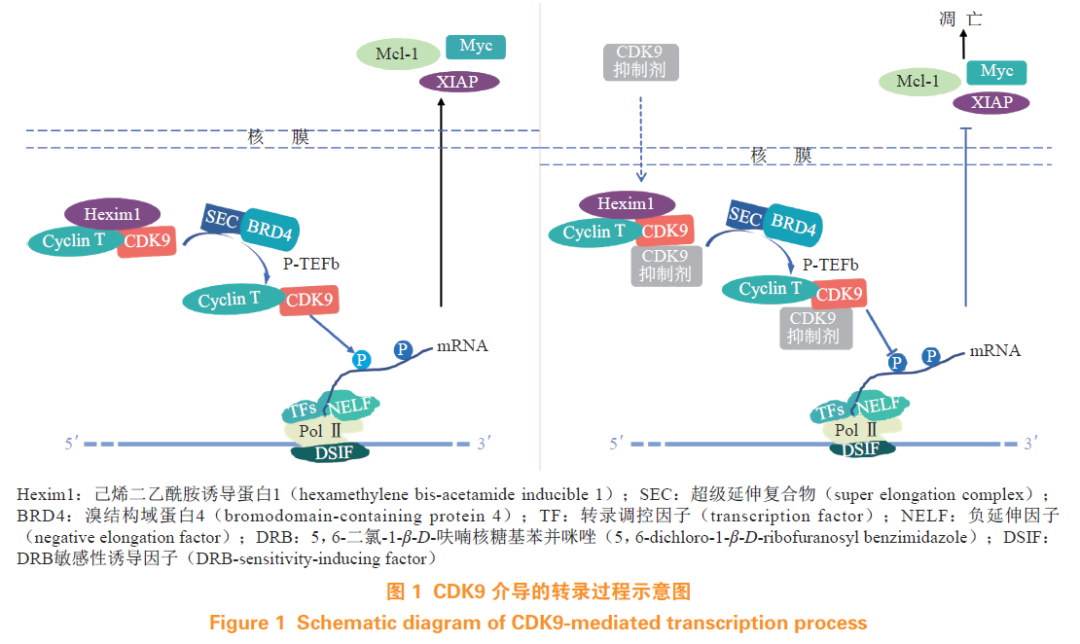
2.2肝细胞癌
从全球的癌症死亡率来看,肝细胞癌(hepatocellular carcinoma,HCC)排在第4位。原发性肝癌类型中,最常见的是肝细胞癌,其主要的危险因素是肝硬化,常由慢性病毒性肝炎、酒精滥用和非酒精性肝病引起。在晚期HCC的患者中,尽管使用多激酶抑制剂进行靶向治疗,也只能将预期寿命从8个月延长到11个月。在HCC发病的发病机制研究中,发现Myc癌蛋白是HCC中的驱动因素之一,Myc的过表达可以诱导异常增殖。在小鼠肝癌模型中,抑制Myc的表达会诱导肿瘤的退化。Myc在DNA复制、转录激活、转录延伸中起着重要作用,虽然具体机制尚不清楚。但是研究发现,肝癌中CDK9介导的转录延伸对维持Myc的高表达有着密切联系。
2.3前列腺癌
前列腺癌(prostate cancer,PCa)是男性中最常见的癌症,在疾病的初始阶段具有雄激素依赖性,并响应雄激素剥夺疗法(androgen deprivation therapy,ADT),80%~90%的患者出现明显的临床消退和缓解,但2~3年后癌症会再次复发,并发展成为去势抵抗性前列腺癌(CRPC),此阶段ADT疗效较差。最终,癌细胞会转移到其他器官,如骨骼、肺、脑和肝脏。疾病进入到晚期阶段,患者的预后较差。雄激素受体(androgen receptor,AR)是PCa存活和进展的关键转录因子。在一般条件下,AR被激活并在雄激素诱导后形成同源二聚体,AR的同源二聚体会在靶基因的启动子区域结合抗氧化反应元件(anti-oxidant response element,ARE),从而介导转录。当PCa进展为CRPC后通过持续激活AR才能在雄激素的去势水平中存活。去势抵抗机制可大致分为3个途径:雄激素依赖性、雄激素非依赖性和旁路。其中旁路途径由抗凋亡蛋白如Bcl-2和Mcl-1的上调介导。CDK9可以调控抗凋亡蛋白的表达,与许多转录因子相互作用并调节它们的活性,其中包括AR。在治疗PCa的过程中AR是众所周知的分子靶标。CDK9可以磷酸化AR结构域上第81位的丝氨酸(Ser81),激活下游通路。尽管CDK1也可以磷酸化Ser81,但其在AR转录活性调节中的功能仍值得怀疑。利用CDK9介导的磷酸化作用调节AR的催化特性。通过抑制CDK9可以限制AR的激活,并抑制疾病进展至CRPC期。研究表明,在小鼠异种移植模型研究和Ⅰ期临床试验中均发现CDK9抑制剂具有抗肿瘤活性。
3具有抗肿瘤活性的CDK9小分子抑制剂
近年来,通过分子模型设计的CDK9抑制剂在体外显示出良好的抗肿瘤活性。CDK9抑制剂按照化学结构主要可以分为:黄酮类、吡唑并嘧啶类、嘌呤类和氨基噻唑类。
3.1黄酮类
3.1.1夫拉平度
天然黄酮类化合物夫拉平度(flavopiridol)是第1个进入临床试验的CDK抑制剂。该化合物的盐酸盐形式(alvocidib hydrochloride)目前处于Ⅱ期临床研究。在2007年,因治疗慢性淋巴细胞白血病(chronic lymphocytic leukemia,CLL)而获得欧盟孤儿药资格;2014年,因治疗AML而在美国获孤儿药资格;2015年,因相同的适应证在欧盟获孤儿药资格。研究证明,夫拉平度可以通过抑制CDK9的活性和减少PolⅡ的磷酸化形式,进而促进细胞凋亡。与其他癌症相比,AML一直缺乏有效的治疗药物,并且药物开发缓慢。在临床试验中发现当以定时序贯疗法与阿糖胞苷和米托蒽醌组合时,夫拉平度在AML中表现出稳定的疗效。在405例AML患者中以定时序贯方案(夫拉平度与米托蒽醌合用,并持续输注阿糖胞苷)进行研究,发现复发和难治性AML的完全缓解率为36%,低风险的AML中,完全缓解率为68%。另一项临床研究显示,夫拉平度能降低Mcl-1的表达。同时在对AML患者使用夫拉平度结合阿糖胞苷或米托蒽醌的Ⅱ期随机研究中,58%的AML患者获得了完全应答,但有8%的患者因不良反应退出研究,13%患者死亡。在CLL的Ⅰ期研究中发现,42例患者中,有19例获得部分应答;中位应答持续时间为12个月。12例17p13染色体缺失患者中有5例获得部分缓解,18例11q22染色体缺失患者中有13例获得部分缓解。在临床试验中发生了不良反应,如血小板减少症、栓塞、中性粒细胞减少症等。
3.1.2Voruciclibhydrochloride
CDK9抑制剂voruciclib hydrochloride是一种小分子黄酮类衍生物,由Piramal Life Sciences研发,目前处于Ⅰ期临床研究阶段。该化合物在结构上类似于夫拉平度,但对CDK9具有更好的选择性,可以减少非预期的脱靶介导的副作用。弥漫性大B细胞淋巴瘤(diffuse large B-cell lymphoma,DLBCL)是常见的非霍奇金淋巴瘤之一,通常对R-CHOP方案(利妥昔单抗联合环磷酰胺、多柔吡星、长春新碱和泼尼松)有应答。然而,仍有约40%的患者继续表现出对化疗耐药的DLBCL或复发,并最终死于该疾病。Voruciclib hydrochloride在多种DLBCL模型中可抑制Mcl-1的表达,且与Bcl-2特异性抑制剂venetoclax联用可诱导肿瘤细胞死亡、抑制肿瘤生长,实现协同的抗肿瘤功效,包括高风险的激活B细胞亚型(active B cell,ABC)。
3.2吡唑并嘧啶类
3.2.1Dinaciclib Dinaciclib是一种小分子CDK抑制剂,其对CDK1、CDK2、CDK5及CDK9均有抑制活性。Dinaciclib由默沙东公司研发,目前处于Ⅲ期临床研究,用于治疗难治愈的CLL。2011年,FDA授予dinaciclib治疗CLL的孤儿药资格。在骨髓瘤治疗方面,dinaciclib是单独使用就可发挥疗效的药物之一,而同类其他许多药物需联用。与夫拉平度比,dinaciclib具有更高的抗骨髓瘤活性和治疗指数,并被证明对CLL具有良好的临床疗效。一项由CLL患者参加的临床研究显示,接受dinaciclib治疗的52例患者其总体应答率为54%;25例17p13.1染色体缺失的患者中,有19例获得应答;而21例没有这种异常的患者中有12例获得应答。难治性患者在接受dinaciclib治疗后并未出现常见的感染、疲劳和腹泻。尽管17p13.1染色体缺失的患者比例高达45%,但应答率仍有54%,并且中位无进展生存期接近1年。与夫拉平度相比,dinaciclib可以长期给药,并对大多数难治性患者有效。
3.2.2BS194BS194对CDK2、CDK5及CDK9的抑制活性的IC50值达纳摩尔级别。药动学研究表明,本品可口服给药,且能抑制CDK底物的磷酸化和异种移植瘤的生长,有望成为具有临床疗效的CDK抑制剂。
3.3嘌呤类
3.3.1SeliciclibSeliciclib是一种口服有效的CDK2、CDK7和CDK9抑制剂,该化合物由法国国家科学研究院、ManRos Therapeutics与Cyclacel共同研发,目前处于Ⅱ期临床研究阶段。Seliciclib在鼻咽癌(nasopharyngeal carcinoma,NPC)和非小细胞肺癌(non-small-cell lung carcinoma,NSCLC)的Ⅰ期和Ⅱ期临床研究中作为单一用药显示出临床疗效。在鼻咽癌的Ⅰ期临床研究中,某些患者在治疗后出现与肿瘤细胞增殖和生存相关的基因转录下调。在Ⅰ期临床研究中,有77例不同实体瘤患者在多次治疗方案失败后接受seliciclib治疗。其中有10名患者病情稳定,至少持续4个月。同时还有2名晚期NSCLC患者在4种治疗方案失败后,病情保持稳定,分别持续14和18个月。
3.3.2CR8CR8是seliciclib的衍生物,其在抑制CDK9、抑制PolⅡ的磷酸化和诱导凋亡方面的活性均要强于seliciclib,但是CR8的毒性和抗肿瘤活性还需要在动物模型中进行进一步研究。
3.4氨基噻唑类
3.4.1SNS-032SNS-032是一类具有氨基噻唑类结构的化合物,可选择性抑制CDK2、CDK7和CDK9。研究显示,其能在体外有效抑制CLL,其机制是抑制PolⅡ的活性,进而减少相应RNA的合成。
3.4.2THAL-SNS-032THAL-SNS-032是一种选择性CDK9降解剂,由SNS-032与沙利度胺拼合形成。目前传统的CDK抑制剂是可逆的且需要连续地占据靶点,而由于ATP结合口袋的同源性,开发高选择性的CDK抑制剂更具有挑战性。THAL-SNS-032以Cereblon蛋白(CRBN)依赖性方式选择性诱导CDK9降解,实现不可逆性抑制细胞转录,进而发挥更有效的抗肿瘤作用。与传统的CDK9抑制剂相比,THAL-SNS-032具有更稳定和持久的药效。
在实验中发现,虽然THAL-SNS-032与另2种传统CDK9抑制剂NVP-2和SNS-032都可以迅速诱导细胞凋亡,但洗脱去除药物后,只有接受过THAL-SNS-032处理的细胞仍处于增殖抑制状态,这可能是CDK9被降解的结果。因此,THALSNS-032比传统的CDK9抑制剂更具有治疗优势。这种将底物降解的策略有助于开发出活性和选择性良好的其他抑制剂(例如以组蛋白乙酰转移酶、组蛋白脱乙酰酶、脱氢酶和磷酸酶等为底物的抑制剂)。虽然降解具有更持久的药理学作用,但是传统抑制剂起效速度更具有优势,因此需要进一步研究THAL-SNS-032的药动学和药效学特性。
4结语与展望
CDK9和细胞周期蛋白结合形成的复合物,对转录过程的调节至关重要,一些研究已经表明CDK9在转录过程中起着重要调节作用。CDK9对维持Mcl-1等短寿命抗凋亡蛋白的表达起着重要作用,并且有一部分肿瘤细胞需要CDK9才能存活。有研究证明,间接调节Mcl-1,可以使肿瘤对BclxL的直接抑制剂敏感。由于Mcl-1对CDK9功能的依赖,并且有报道证明CDK抑制剂和Bcl-2同源结构域3(Bcl-2 homology domain3,BH3)拟合物之间存在着协同作用。这使得以CDK9为靶点的药物更具有研究价值。
目前为止,已经有多个CDK小分子抑制剂处于临床研究阶段,某些抑制剂通过与传统化学疗法联用产生了较好的疗效。但是CDK家族成员众多,它们之间的结构相似性、在不同肿瘤之间的表达差异以及CDK9与细胞周期蛋白所组成的复合物的具体功能等基础研究尚未完全清楚,使得现有的CDK9抑制剂存在选择性差、脱靶等问题。因此,寻找具有高特异性的CDK9抑制剂,并探索CDK9抑制剂与其他药物联用的可能性及相关策略,有望推动CDK9抑制剂在抗肿瘤治疗方面的进展。
关于药学进展
感谢您阅读《药学进展》微信平台原创好文,也欢迎各位读者转载、引用。本文选自《药学进展》2020年第3期。
《药学进展》杂志是由中国药科大学和中国药学会共同主办、国家教育部主管,月刊,80页,全彩印刷。刊物以反映药学科研领域的新方法、新成果、新进展、新趋势为宗旨,以综述、评述、行业发展报告为特色,以药学学科进展、技术进展、新药研发各环节技术信息为重点,是一本专注于医药科技前沿与产业动态的专业媒体。
《药学进展》注重内容策划、加强组稿约稿、深度挖掘、分析药学信息资源、在药学学科进展、科研思路方法、靶点机制探讨、新药研发报告、临床用药分析、国际医药前沿等方面初具特色;特别是医药信息内容以科学前沿与国家战略需求相合,更加突出前瞻性、权威性、时效性、新颖性、系统性、实战性。根据最新统计数据,刊物篇均下载率连续三年蝉联我国医药期刊榜首,复合影响因子0.760,具有较高的影响力
来源:ppsyxjz 药学进展
原文链接:https://mp.weixin.qq.com/s?__biz=MzA5MDY3ODExNQ==&mid=2651310431&idx=1&sn=70ab7e3b8f032be355b6c961ede50009&chksm=8bf49f53bc831645c75bbdf7c4d79a26ff93ea91e3de26f611ab6fa05e4914b912ab6ab1971b#rd
版权声明:除非特别注明,本站所载内容来源于互联网、微信公众号等公开渠道,不代表本站观点,仅供参考、交流、公益传播之目的。转载的稿件版权归原作者或机构所有,如有侵权,请联系删除。
电话:(010)86409582
邮箱:kejie@scimall.org.cn


